吴云东课题组在蛋白质折叠模拟领域取得重要进展
时间:2014-07-02
吴云东课题组在蛋白质折叠模拟领域取得重要进展
大部分蛋白质分子在合成出来以后,能在适当的条件下自发地折叠形成特定的三维结构,从而实现其生物学功能。如果能用计算机模拟蛋白质折叠的过程,不仅可以预测其天然态结构,而且可以研究这种神奇的自发有序化的机理。然而,蛋白质折叠的分子动力学(MD)模拟仍然是个挑战,需要准确的物理模型(力场)和足够长的计算时间。近年来,D.E. Shaw研究中心投入巨资设计制造了专门用于MD模拟的超级计算机Aton,利用他们开发的Charmm22*力场,不利用任何已知的蛋白质结构信息,实现了对12个快速折叠的蛋白质分子的折叠模拟(Science 2011)。但近年来并没有课题组利用通常能获得的计算资源成功实现大规模的从头折叠模拟。
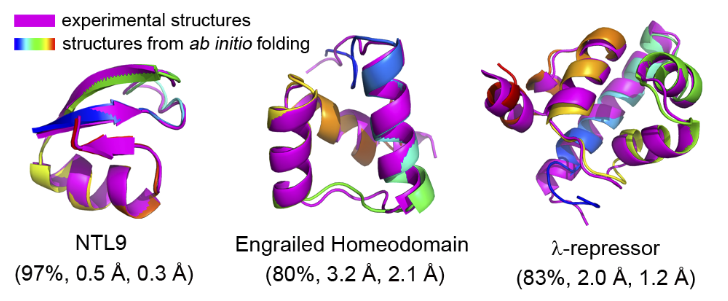
吴云东教授课题组使用本课题组自主开发的残基特异力场(RSFF1),并利用副本交换分子动力学(REMD)模拟来提高构象取样的效率,成功折叠了这12个蛋白质。另外还折叠了两个Charmm22*力场不能很好稳定的蛋白质。此研究表明了RSFF1力场的优势,以及REMD方法的效率。在模拟中还发现蛋白在非折叠态就已经存在相当多的二级结构,另外还发现折叠过程的“摩擦”约为2.5 kcal/mol。该成果以通讯(communication)形式发表在近期的《美国化学会志》上。
文章链接:http://pubs.acs.org/doi/abs/10.1021/ja502735c
该文章由我院的吴云东教授和蒋帆博士共同完成,得到了国家自然科学基金(21133002, 21203004),深圳市孔雀计划项目,以及深圳市重点实验室项目(计算化学与药物设计)的支持。