Li Li, Feifei Song, Xiumei Zhong, Yun-Dong Wu, Xinhao Zhang,* Jiean Chen,* and Yong Huang.* Adv. Synth. Catal. 2019, 361, 1–8
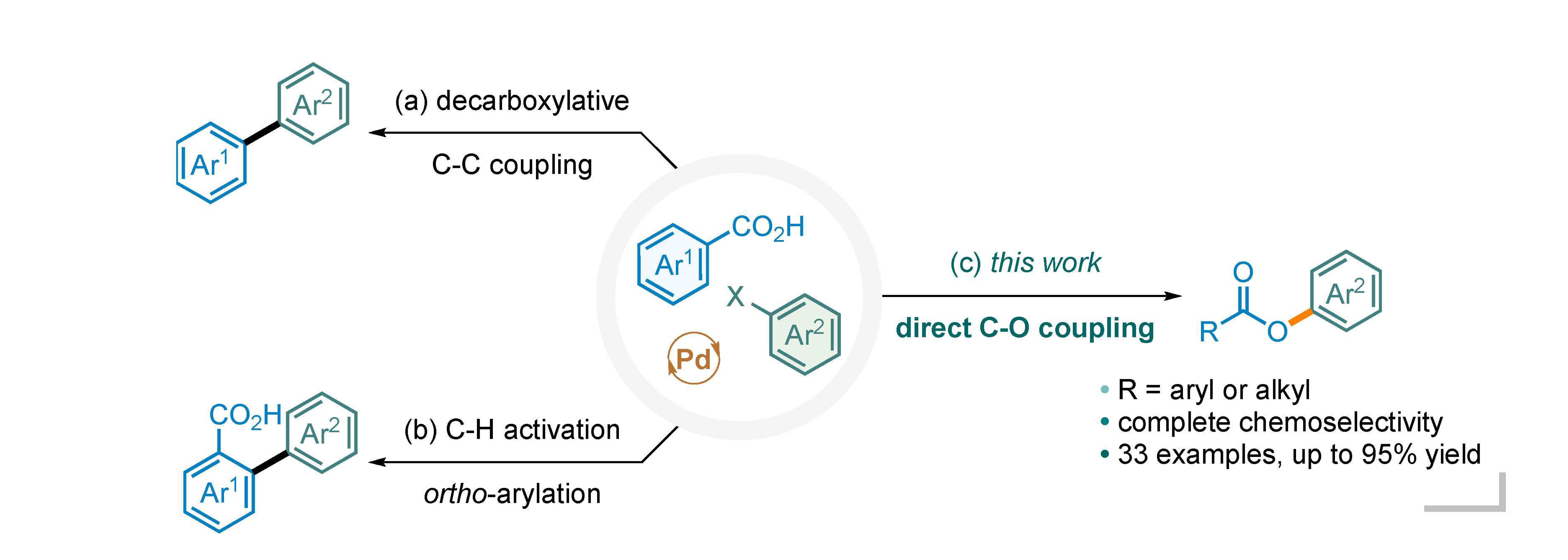
Palladium-catalyzed cross-coupling reactions between carboxylic acids and aryl halides have several possible competitive pathways. Decarboxylative C-C bond coupling and C-H arylation are well established in the literature. However, direct C-O bond coupling between carboxylic acids and aryl halides has received little success. In this report, we describe a protocol for exclusive C-O bond formation, enabled by a bidentate N,N-ligand such as 1,10-phenanthroline. The reaction is general for a broad range of carboxylic acids and iodoarenes. Experimental evidence and computational results suggest a high energy barrier for the alternative pathway of decarboxylative carbon-carbon bond coupling.
59. Alcohol-Directed ortho-C–H Alkenylation
Li Li, Qinglan Liu, Jiean Chen *, Yong Huang *. Synlett 2019, 30, A-E. DOI: 10.1055/s-0037-1611538
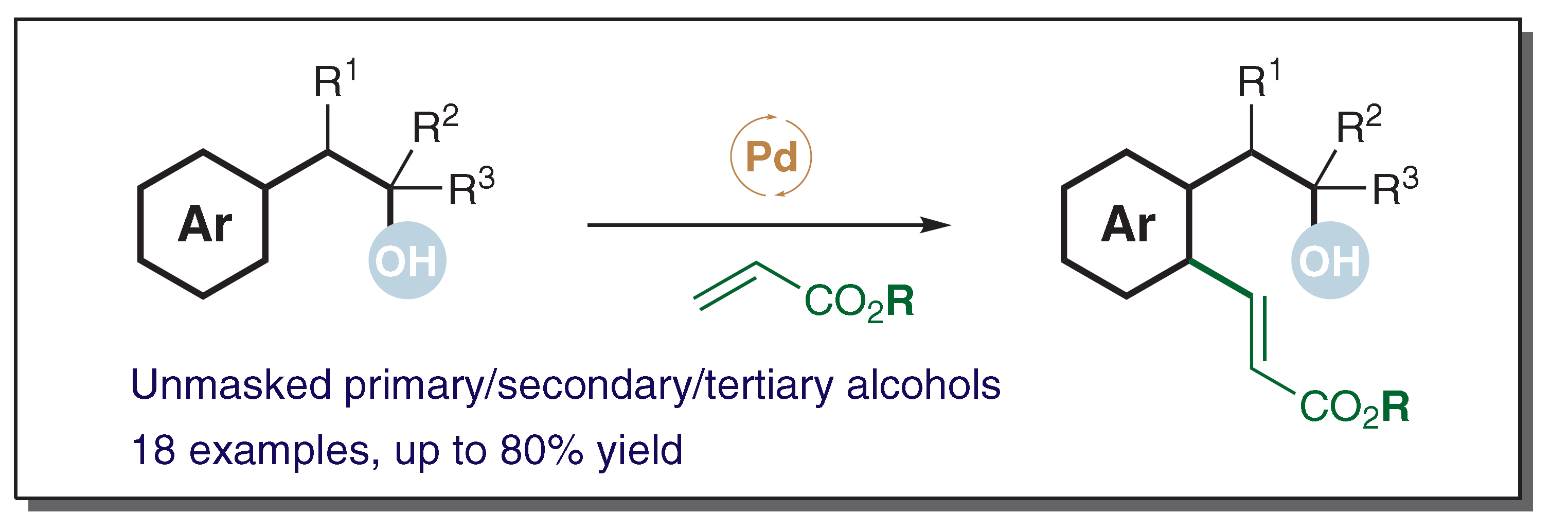
We report a simple and mild dehydrogenative cross-coupling reaction of unprotected arylethanols and acrylates. Unlike the case of previous reactions, in which prior functionalization of the substrate with a metal-coordinating site was required, free primary, secondary, or tertiary hydroxy groups were found to be effective directing groups for the ortho-C–H palladation and subsequent olefination.
58. Enantio- and Diastereoselective Hydrofluorination of Enals via N-Heterocyclic Carbene Catalysis
Leming Wang, Xinhang Jiang, Jiean Chen,* Yong Huang*. Angew. Chem. Int. Ed. 2019. DOI:10.1002/anie.201902989
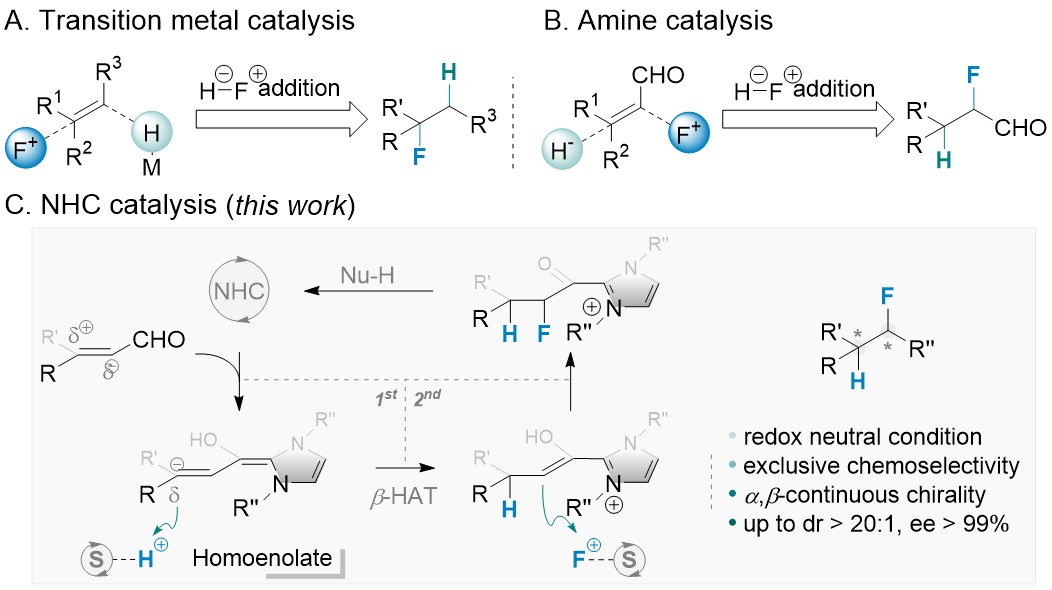
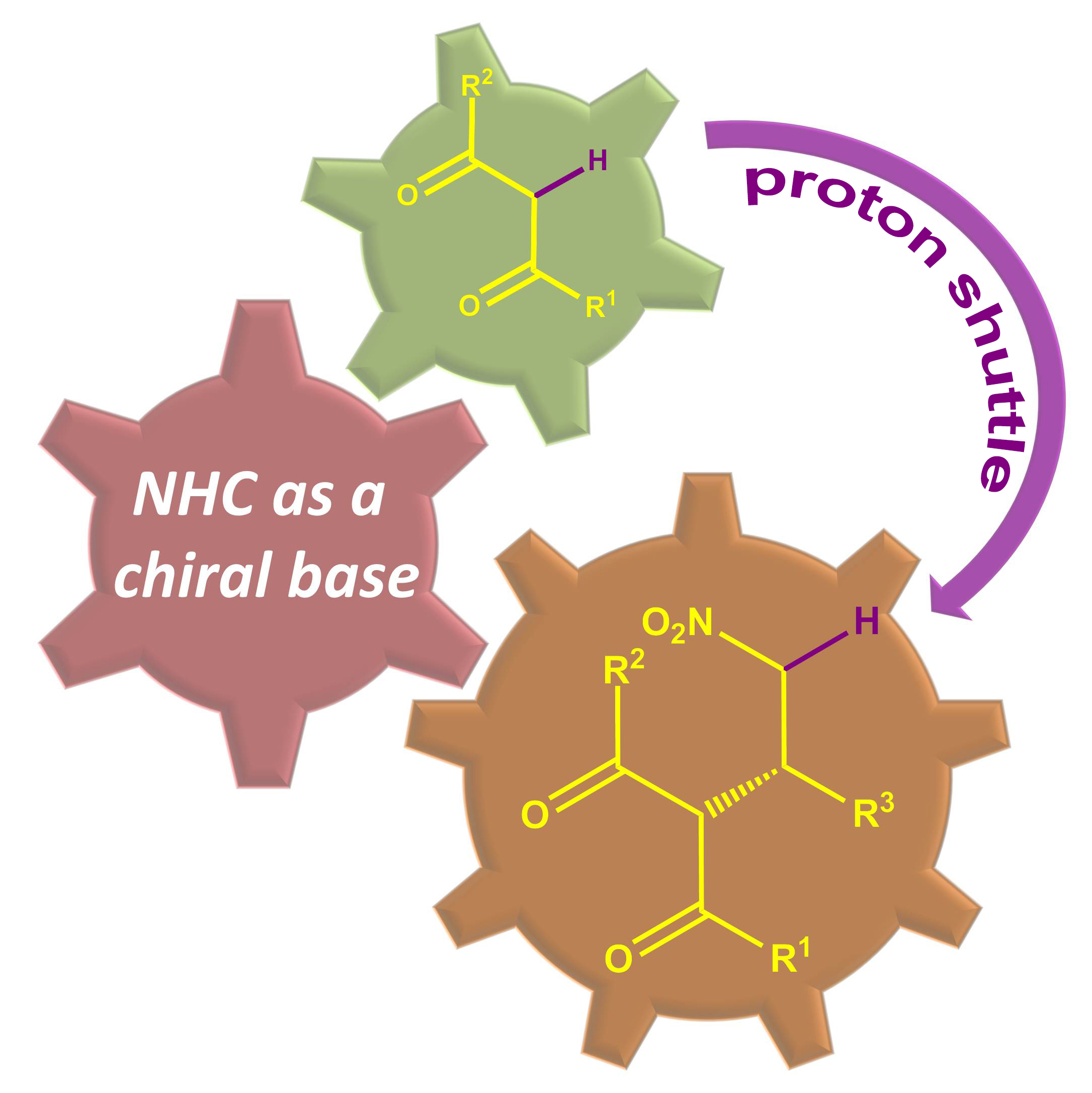
In contrast to well-established asymmetric hydrogenation reactions, enantioselective protonation is an orthogonal approach to create highly valuable methine chiral centers under redox-neutral conditions. We report highly enantio- and diastereoselective hydrofluorination of enals via an asymmetric β-protonation-α-fluorination cascade catalyzed by N-heterocyclic carbene. The two nucleophilic sites of a homoenolate intermediate, generated from enals and an N-heterocyclic carbene, are sequentially protonated and fluorinated. Our results show that controlling the relative rates of protonation, fluorination and esterification is crucial for this transformation, which can be accomplished using a dual shuttling strategy. Structurally diverse carboxylic acid derivatives with two continuous chiral centers are prepared in a single step with excellent dr and ee.
57. N‐Heterocyclic Carbenes as Brønsted Base Catalysts
Jiean Chen, Yong Huang. Book Author(s): Akkattu T. Biju. DOI :10.1002/9783527809042.ch9
This chapter summarizes the recent development of N‐heterocyclic carbenes (NHC) catalysis using noncovalent interactions, with an emphasis on its stereoselective aspects. For reactions involving NHCs as Brønsted base catalysts, the predominant factor concerning the catalytic activity is the pKavalue of the corresponding precatalysts. The types of reactions catalyzed by NHCs behaving as Brønsted bases fall into two categories: transesterifications and conjugate additions. In both cases, strong hydrogen‐bonding interactions between NHCs and nucleophiles accelerate the reactions by elevating the highest occupied molecule orbital (HOMO) energy of the nucleophiles, which react with either esters or electron‐deficient olefins. The role of NHC in transesterification reactions led to further interests in density functional theory (DFT) calculations. The catalytic activity of various NHC catalysts was evaluated using conjugate addition reactions of 1,3‐dicarbonyl compounds to various Michael acceptors. In 2015, Huang and coworkers reported the noncovalent activation of NHC catalysts in the asymmetric sulfa‐Michael addition (SMA).
56. Dehydrogenative Annulation of γ,δ-Unsaturated Amides and Alkynes via Double C-H Activation
Wang Zhen, Li En, He Zhiqi, Chen Jiean, Huang Yong. Acta Phys. -Chim. Sin. 2019, 35, 0001–0009. DOI: 10.3866/PKU.WHXB201811038.
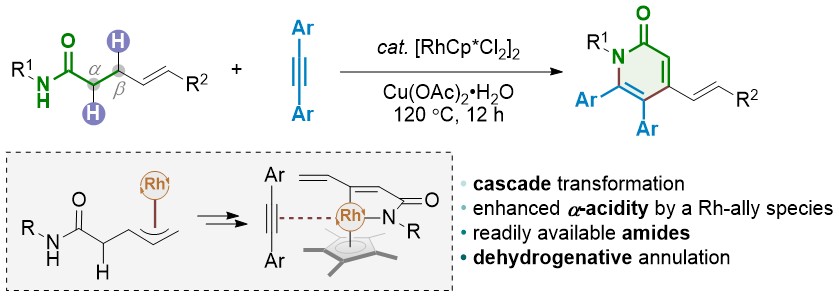
Pyridones represent an important family of heterocycles that exhibit a wide range of biological activities. They are often found in pharmaceutical agents and biomolecules. Several transition-metal-catalyzed transformations have been developed to access this family of heterocycles. Among them, C-H bond activation has recently emerged as a general strategy for the construction of substituted pyridones. In most cases, the core nitrogen-containing heterocycle is assembled via the dehydrogenative annulation of α,β-unsaturated amides and alkynes. Such processes involve a cascade sequence of N-H cleavage, sp2 C-H activation, and annulation. Despite this progress, the more readily available α,β-saturated amides are rarely used. Ideally, tethering the direct dehydrogenation of an amide with the above-mentioned C-H annulation cascade would give a more practical synthesis of pyridones. Nevertheless, the dehydrogenation of amides under mild conditions is a synthetic challenge due to their intrinsic weak α-acidity. Recently, we have reported a general protocol for the aerobic dehydrogenation of γ,δ-unsaturated amides, acids, and ketones. A key Ir-allyl intermediate was believed responsible for enhancing the α-acidity of the amides studied, which enables the dehydrogenation step to occur under mild reaction conditions. Herein, we describe a new method for the synthesis of polysubstituted pyridones using γ,δ-unsaturated amides and alkynes. In the presence of[RhCp*Cl2]2, the dehydrogenation step occurs via β-C-H bond activation. The resulting π-allyl-Rh intermediate undergoes an accelerated dehydrogenation reaction to afford the doubly unsaturated amide. This in-situ generated dienamide undergoes sp2 C-H activation at the β-position and a subsequent alkyne insertion/cyclization reaction to yield the target heterocycle. Regeneration of the Rh catalyst is accomplished using an external oxidant and completes the streamlined double C-H activation and double dehydrogenation catalytic cycle. Various functional groups are well tolerated. The γ-alkenyl moiety not only facilitates the direct dehydrogenation of amides, but also serves as a handle for further derivatization of the as-obtained products. To gain a mechanistic insight into the reaction cascade, a set of control experiments were carried out. The results demonstrate that the dienamide is one of the key reaction intermediates. NMR experiments confirmed that the fast dehydrogenation process occurs during the early stage of the reaction. The alkyne insertion is believed to be the rate-determining step in the reaction cascade, as suggested by competition experiments.
55.Switching Reaction Pathways by Cooperative Catalysis of N-Heterocyclic Carbene and Lewis Acids
Wang Leming, Wang Qian, Chen Jiean*, Huang Yong*. Acta Chim. Sinica 2018, 76, 850—856.
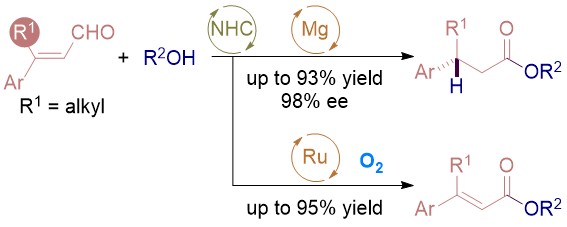
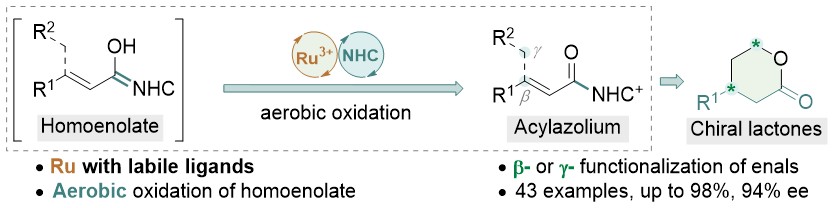
The combination of N-heterocyclic carbenes (NHCs) and Lewis acids (LA) have been occasionally employed in asymmetric annulation reactions. However, synergistic effect of LA on NHC-mediated reactions remains scarce. Herein, we demonstrate that by switching LA co-catalysts, two distinct active species, homoenolate and acyl azolium, can be accessed from the same set of substrates. NHC-catalyzed enantioselective hydroesterification is one of the most straightforward strategies to prepare β-chiral esters. Despite recent advances for this redox-neutral transformation, obtaining high enantioselectivity and yield remains challenging. We recently reported synergistic catalysis, combining an achiral NHC and a chiral phosphoric acid, enables highly enantioselective hydrothioesterification and hydroesterification of enals. However, both stereoselectivity and yield for hydroesterification are far from ideal. Specifically, sluggish reactions, accompanied with ee’s in mid-80% are often obtained. Additionally, competing pathways for E/Z isomerization and oxidative esterification of enal are serious for a number of substrates. In order to address this issue, we propose a new cooperative catalytic system, consisting of a NHC, a LA and a proton-shuttling agent, might accelerate the pivotal asymmetric β-protonation process. We suspect that the choice of LA might provide complementary reaction pathways from the same enal substrates. Starting from β-alkyl cinnamaldehydes, highly enantioselective hydroesterification is accomplished via asymmetric β-protonation enabled by a magnesium co-catalyst. In sharp contrast, the same homoenolate intermediate can undergo aerobic oxidation, via single electron transfer (SET), in the presence of a ruthenium co-catalyst. Control experiments show distinct rate difference between the E- and Z-isomers of enal. Substrates with Z-configuration react significantly slower under the standard reactions. E/Z isomerization is also slow. Photoirradiation was applied to address the challenging issue of isomeric enals and both high yield and ee are obtained using start materials as E/Z mixtures.

54. Structure-Based Drug Design and Identification of H2O‑Soluble and Low Toxic Hexacyclic Camptothecin Derivatives with Improved Efficacy in Cancer and Lethal Inflammation Models in Vivo
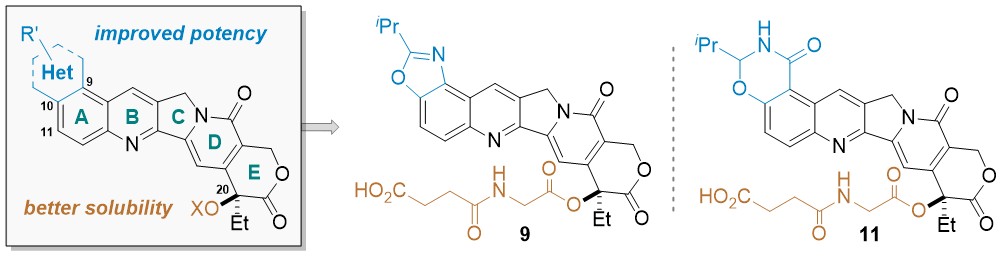
Peichen Pan,† Jiean Chen,§ Xijian Li,§ Miyang Li,§ Huidong Yu,∥ Jean J. Zhao,⊥,# Jing Ni,⊥,#Xuwen Wang,† Huiyong Sun,† Sheng Tian,∇ Feng Zhu,† Feng Liu,∇ Yong Huang,*,§ and Tingjun Hou*,†,‡ J. Med. Chem. 2018, XXX, XXX−XXX. DOI: 10.1021/acs.jmedchem.8b00498
Camptothecin (CPT) has been shown to block disassembly of the topoisomerase I (Topo I)/DNA cleavable complex. However, the poor aqueous solubility, intrinsic instability, and severe toxicity of CPTs have limited their clinical applications. Herein, we report the design and synthesis of H2O-soluble and orally bioavailable hexacyclic CPT derivatives. By analysis of a virtual chemical library and cytotoxicity screening in vitro, 9 and 11 were identified as potential prodrugs and chosen for further characterization in vivo. Both compounds exhibited remarkable anticancer and anti-inflammation efficacies in animals and improved drug-like profiles.
53. Aerobic Oxidation/Annulation Cascades via Synergistic Catalysis of RuCl3 and N‐Heterocyclic Carbenes
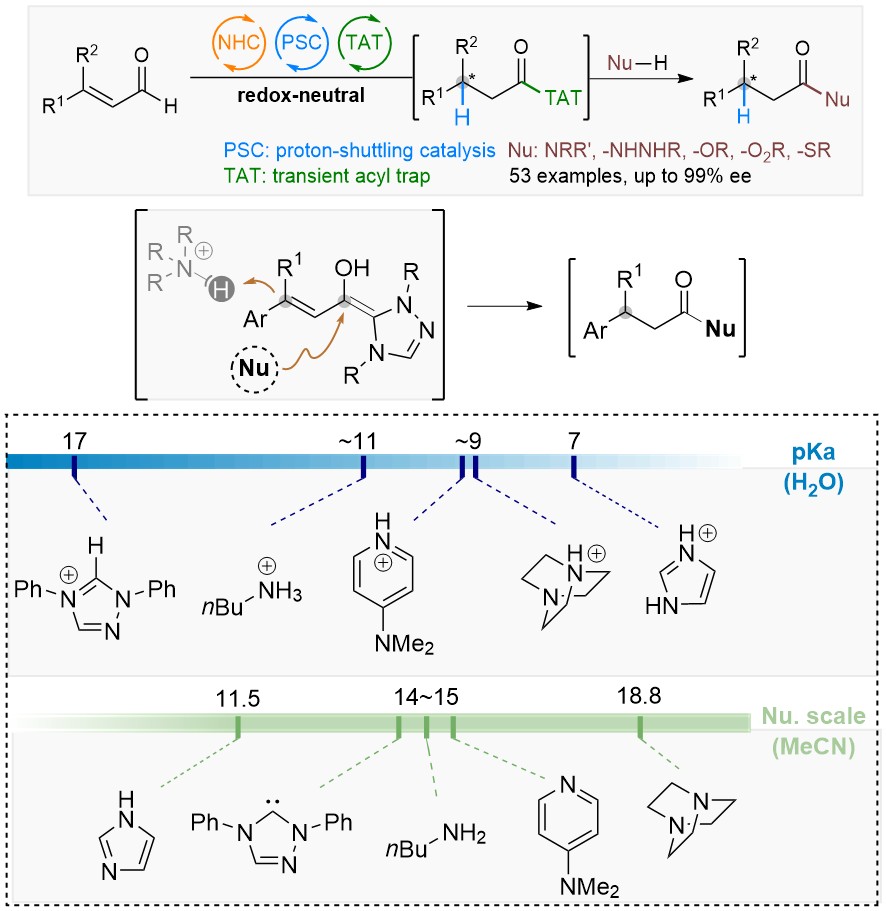
52. Enantioselective hydroamidation of enals by trapping of a transient acyl species
Pengfei Yuan, Jiean Chen*, Jing Zhao*, Yong Huang*. Angew. Chem. Int. Ed. 2018. 57, 8503 –8507. DOI: 10.1002/anie.201803556
An enantioselective synthesis of β-chiral amides by asymmetric and redox-neutral hydroamidation of enals is reported. In this reaction, a chiral N-heterocyclic carbene (NHC) catalyst reacts with enals to generate the homoenolate intermediate. Upon highly enantioselective β-protonation via proton-shuttle catalysis, the resulting azolium intermediate reacts with imidazole to yield the key β-chiral acyl species. This transient intermediate provides access to diversified β-chiral carbonyl derivatives, such as amides, hydrazides, acids, esters, and thioesters. In particular, β-chiral amides can be prepared in excellent yield and ee (40 chiral amides, up to 95% yield and 99% ee). This modular strategy overcomes the challenge of the disruption by basic amines of the high selective proton-shuttling process.
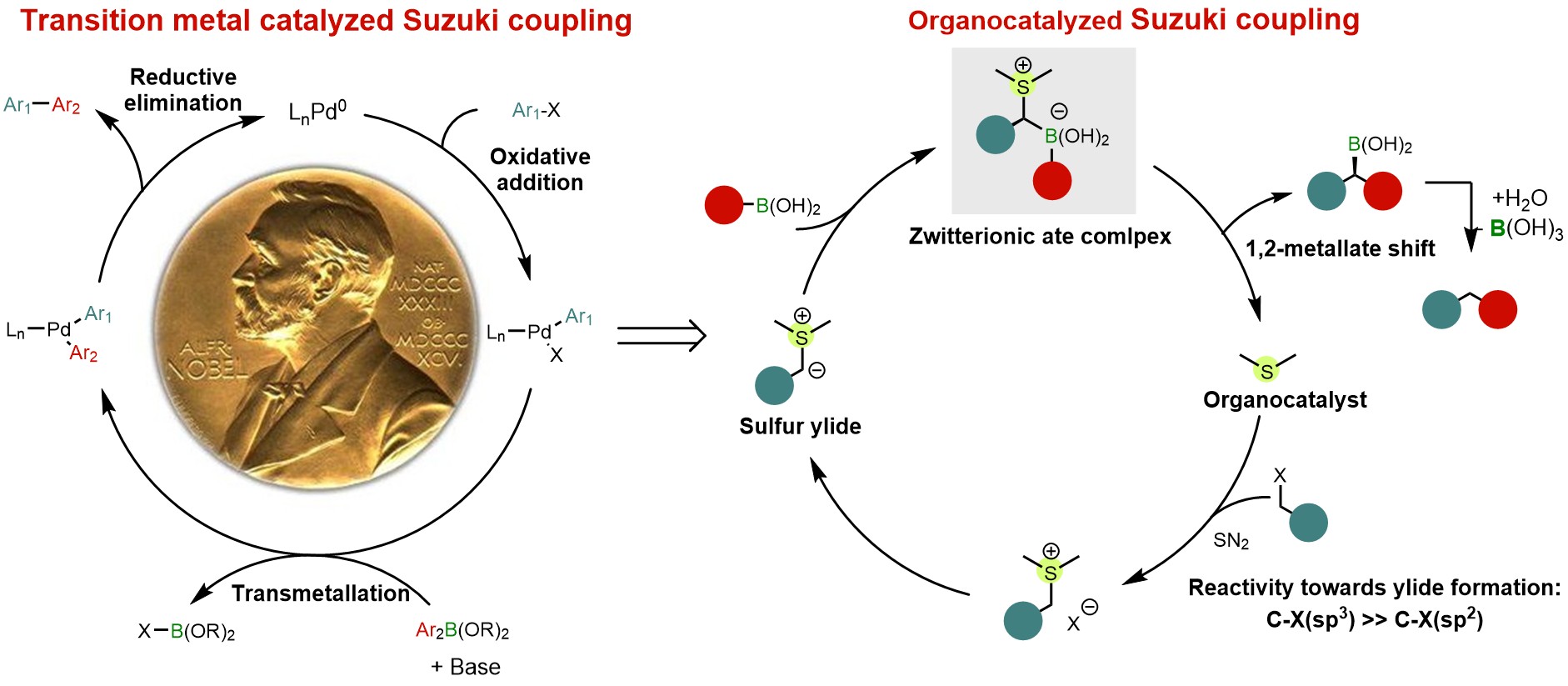
Zhiqi He, Feifei Song, Huan Sun, and Yong Huang*. J. Am. Chem. Soc. 2018, 140, 2693-2699. DOI: 10.1021/jacs.8b00380
Cross-coupling of organoboron compounds with electrophiles (Suzuki-Miyaura reaction) have greatly advanced C-C bond formation and well received in medicinal chemistry. During the last 50 years, transition metals play a central role throughout the catalytic cycle of this im-portant transformation. In this process, chemoselectivity among multiple carbon-halogen bonds is a familiar challenge. In particular, selective oxidative addition of transition metals to alkyl halides rather than aryl halides is difficult due to unfavorable transition states and bond strengths. We describe a new approach that uses a single organic sulfide catalyst to activate both C(sp3)-halides and aryl boronic acids via a zwitterionic boron “ate” intermediate. This “ate” species undergoes a 1,2-metallate shift to afford Suzuki coupling products using benzyl chlo-rides and aryl boronic acids. Various diaryl methane analogues can be prepared, including those with complex and biologically active motifs. The reactions proceed under transition-metal-free conditions and C(sp2)-halides, including aryl bromides and iodides, are unaffected. The orthogonal chemoselectivity is demonstrated in the streamlined synthesis of highly functionalized diaryl methane scaffolds using multi-halogenated substrates. Preliminary mechanistic experiments suggest both the sulfonium salt and the sulfur ylide are involved in the reaction, with the formation of sulfonium salt being the slowest step in the overall catalytic cycle.
50. Streamlined asymmetric α-difunctionalization of ynones
Siyu Peng1, Zhaofeng Wang1, Linxing Zhang, Xinhao Zhang* & Yong Huang*. Nat. Commun. 2018, 9, 375. DOI: 10.1038/s41467-017-02801-9
Ynones are a unique class of structural motifs that show remarkable chemical versatility. Chiral ynones, particularly those possessing an α-stereogenic center, are highly attractive templates for structural diversification. So far, only very limited examples have been reported for asymmetric α-functionalization of ynones. Asymmetric double α-functionalization of ynones remains elusive. Here we describe a streamlined strategy for asymmetric α- difunctionalization of ynones. We developed a gold-catalyzed multicomponent condensation reaction from a simple ynone, an amine, and an electrophilic alkynylating reagent to generate a 1,2-dialkynyl enamine, a key stable and isolable intermediate. This intermediate can undergo asymmetric fluorination catalyzed by a chiral phosphoric acid derivative. Chiral ynones with an α-quaternary carbon and containing a fluorine and an alkyne can be synthesized in high yield and high ee. The synthetic utility of this method is demonstrated by the synthesis of enantioenriched tri(hetero)arylmethyl fluorides.

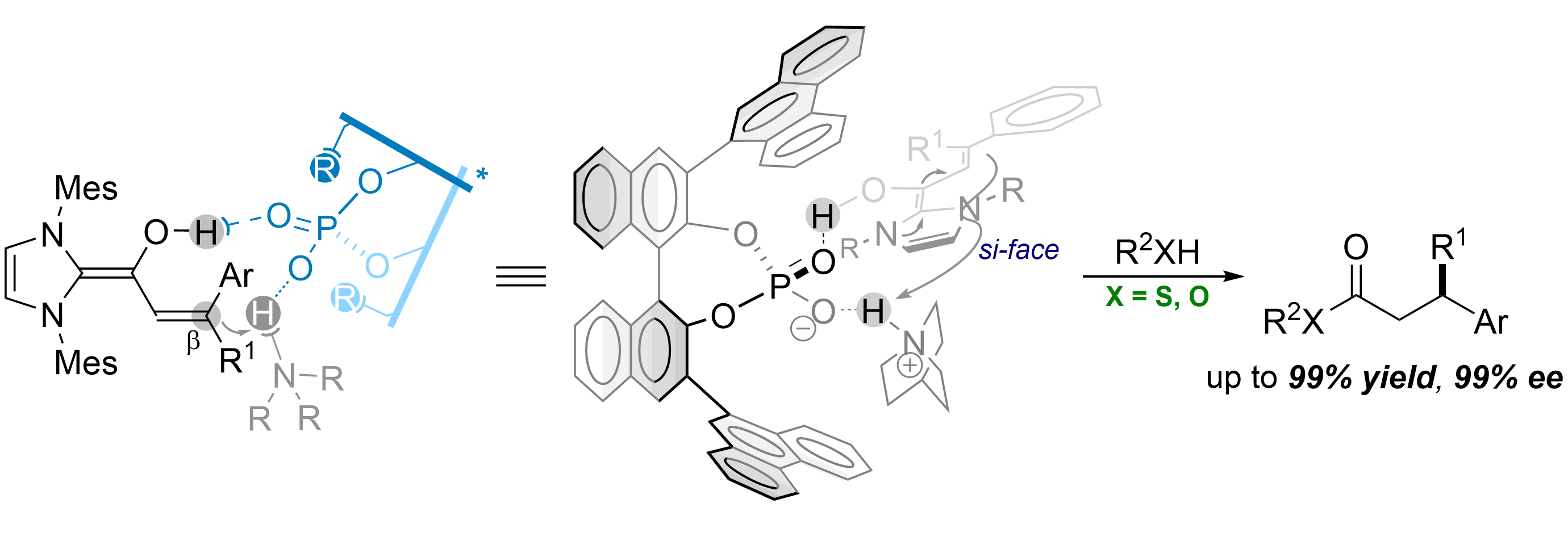
49. Enantioselective Cooperative Proton-Transfer Catalysis using Chiral Ammonium Phosphates
Linrui Zhang, Pengfei Yuan, Jiean Chen* and Yong Huang*. Chem. Commun., 2018, 54, 1473. DOI: 10.1039/C7CC09549J
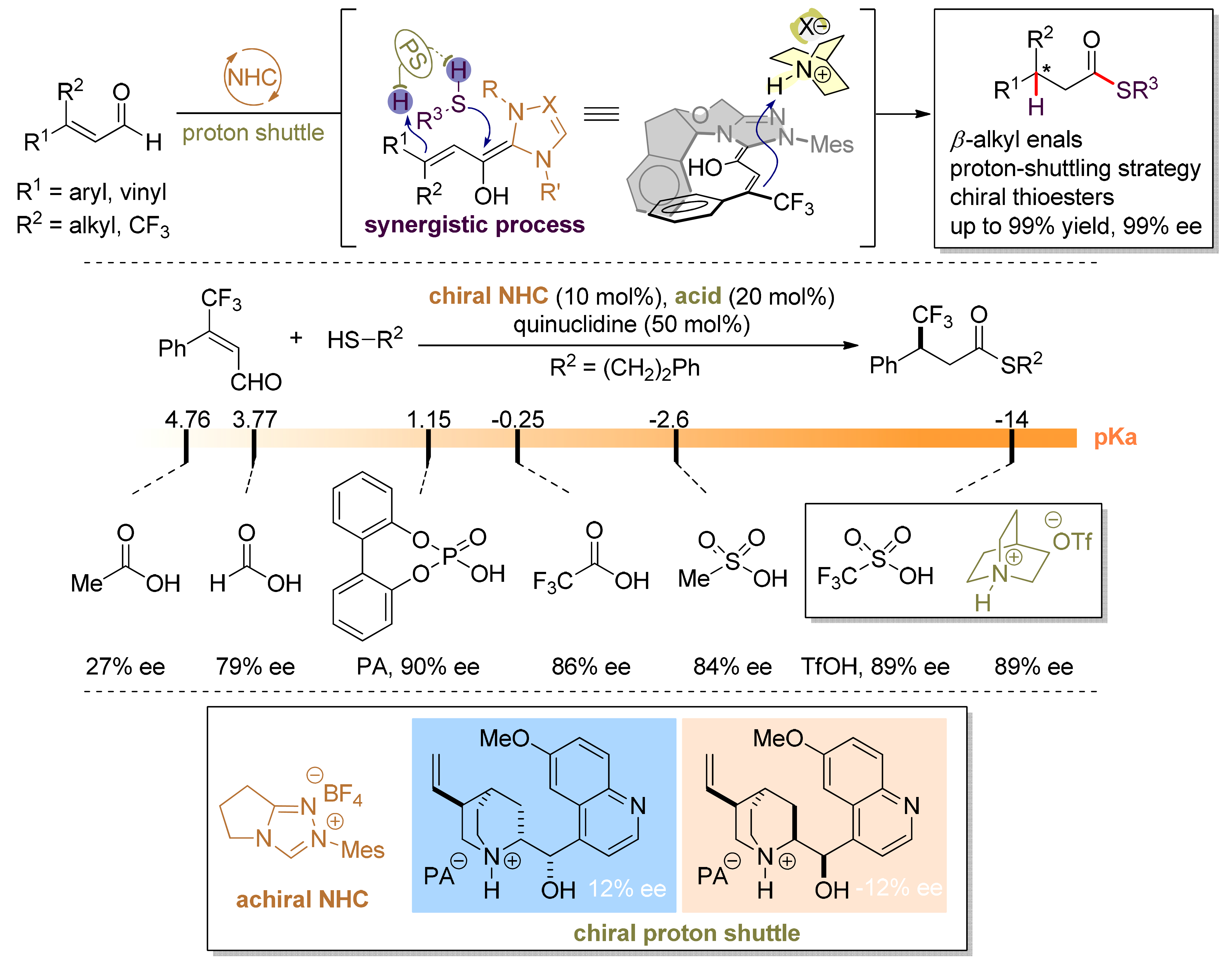
Chiral phosphorate anions are shown to be highly en-antioselective templates for proton-transfer catalysis. A salt generated in situ from a bridgehead amine and a BINOL-derived chiral phosphoric acid serves as an effective proton-shuttle that delivers remarkable enan-tioselectivity in a bioinspired, triple co-operative catal-ysis involving an achiral NHC. Thioesters with a β-chiral center can be prepared in a single step from sub-stituted cinnamaldehyde derivatives, with up to 99% yield and 99% ee. Heteroaryl groups are well tolerated in these reactions, despite the presence of basic sites.
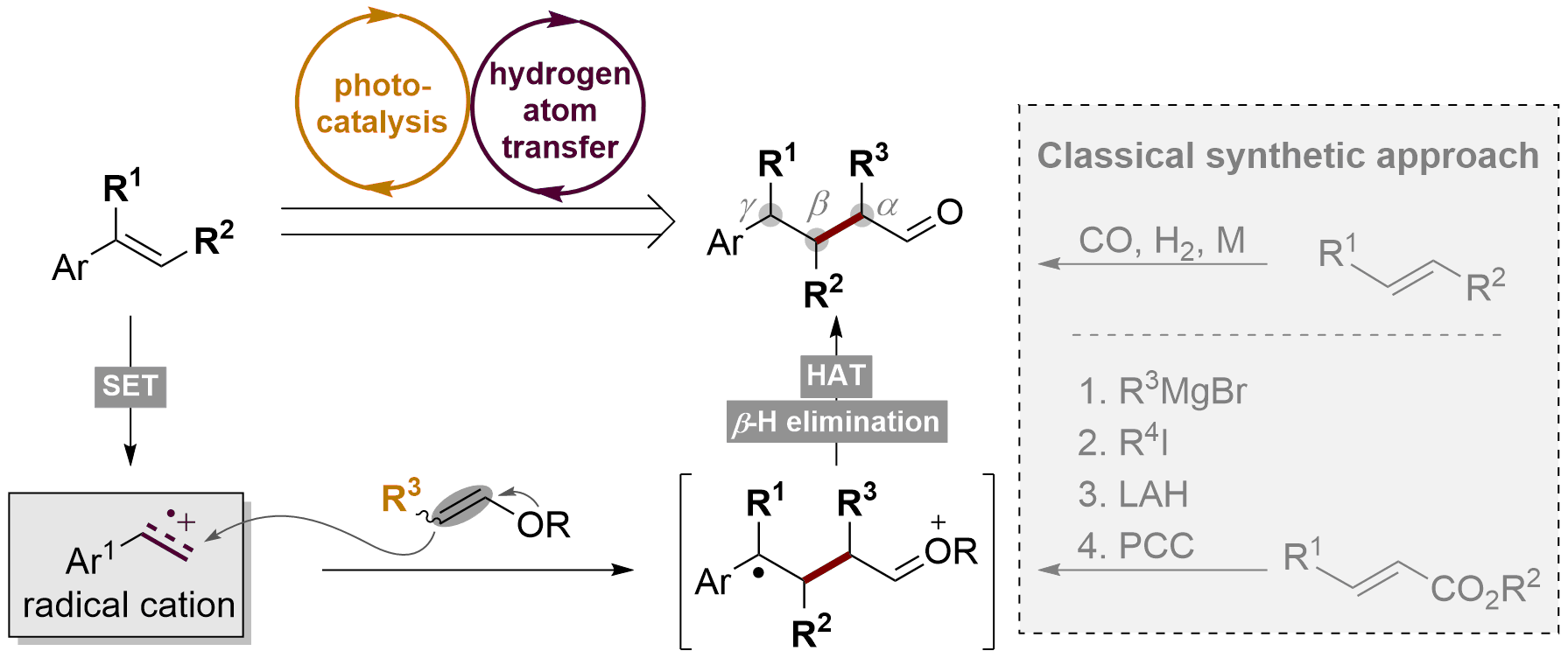
48. Direct Synthesis of Polysubstituted Aldehydes via Visible-Light-Catalysis
Fengjin Wu1, Leifeng Wang1, Jiean Chen*, David A. Nicewicz*, Yong Huang*. Angew. Chem. Int. Ed. 2018, 57, 2174-2178.
Aldehydes are one of the most versatile functional groups for synthetic chemistry. However, access to polysubstituted alkyl aldehydes is very limited and requires lengthy synthetic routes that involve multiple-step functional group interconversion. Herein, we report a one-step synthesis of polysubstituted aldehydes from readily available olefin substrates using visible light photoredox catalysis. Despite a number of competing reaction pathways, commercial styrenes react with vinyl ethers selectively in the presence of an acridinium salt photooxidant and a disulfide hydrogen-atom-transfer catalyst under blue LED irradiation. Alkyl aldehydes with different substitution patterns are prepared in good yields. This strategy can be applied to structurally sophisticated substrates.
Zhen Wang1, Zhiqi He1, Linrui Zhang and Yong Huang*, J. Am. Chem. Soc., 2018, 140, 735−740. DOI: 10.1021/jacs.7b11351
Direct aerobic α,β-dehydrogenation of γ,δ-unsaturated amides and acids using a simple iridium/copper relay catalysis system is described. We developed a new strategy that overcomes the challenging issue associated with the low α-acidity of amides and acids. Instead of α-C–H metalation, this reaction proceeds by β-C–H activation, which results in enhanced α-acidity. Conjugated dienamides and dienoic acids were synthesized in excellent yield with this reaction, which uses a simple reaction protocol. Mechanistic experiments suggest a catalyst resting state mechanism in which both α-C–H and β-C–H cleavage is accelerated.


46. Amine-Triggered 6p-Electrocyclization–Aromatization Cascade of Ynedienamines
Xijian Li, Huidong Yu, and Yong Huang*, Adv. Synth. Catal. 2017, 359, 1379 – 1387
An unprecedented 6p-electrocyclization–aromatization cascade reaction of ynedienamines is described. The electron-rich ynedienamine is converted by g-protonation to an electrophilic allenyl iminium species which is susceptible to amine addition generating a highly electron-rich triene intermediate. The 6p-electrocyclization is accelerated by three electron-donating substituents in a captodative manner. Subsequent redox-neutral aromatization allows the direct synthesis of 1,3-diaminobenzenes from readily available ynedienamines.

Peichen Pan, Huidong Yu, Qinglan Liu, et al. Yong Huang*, and Tingjun Hou*, ACS Cent. Sci. 2017, DOI: 10.1021/acscentsci.7b00419
Targeted inhibition of anaplastic lymphoma kinase (ALK) dramatically improved therapeutic outcomes in the treatment of ALK positive cancers, but unfortunately patients invariably progressed due to acquired resistance mutations in ALK. Currently available drugs are all type-I inhibitors bound to the ATP-binding pocket and are most likely to be resistant in patients harboring genetic mutations surrounding the ATP pocket. To overcome drug resistance, we rationally designed a novel kind of “bridge” inhibitor, which specially bind into an extended hydrophobic back pocket adjacent to the ATP-binding site of ALK. The novel type-I1/2 inhibitors display excellent antiproliferation activity against ALK-positive cancer cells and appear superior to two clinically used drugs, crizotinib and ceritinib. Structural and molecular modeling analyses indicate that the inhibitor induces dramatic conformational transition and stabilizes unique DFG-shifted loop conformation, enabling persistent sensitivity to different genetic mutations in ALK. These data highlight a rationale for further development of next-generation ALK inhibitors to combat drug resistance.
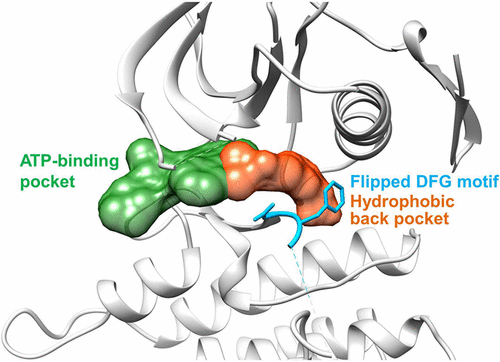
44. Construction of Pyridazine Analogues via Rhodium-mediated C-H Activation
Chao Yang, Feifei Song, Jiean Chen*, and Yong Huang*, Adv. Synth. Catal. 2017, 359, 3496-3502.
Herein a rhodium (III)-mediated catalysis was demonstrated for approaching the structurally divergent N,N-bicyclic pyridazine analogues. The pyrazolidinone moiety was used to direct the ortho C-H activation and this led to a general synthesis of benzopyridazine analogues with satisfactory yields. The crucial effect of the base was illustrated in the sequential dehydration process. For mechanistic insight, control experiments were performed for illustration of the catalytic circle. Gram scale synthesis and several practical transformations were conducted for further applications.

43. Enantioselective β‑Protonation of Enals via a Shuttling Strategy
Jiean Chen1, Pengfei Yuan1, Leming Wang, and Yong Huang*, J. Am. Chem. Soc. 2017, 139, 7045−7051.
Remote asymmetric protonation is a longstanding challenge due to the small size of protons. Reactions involving electron-deficient olefins pose a further difficulty due to the electrophilic nature of these substrates. We report a shuttling system that delivers a proton in a highly enantioselective manner to the β-carbon of enals using a chiral N-heterocyclic carbene (NHC) catalyst. Choices of a Brønsted base shuttle and a Brønsted acid cocatalyst are critical for highly stereoselective β-protonation of the homoenolate intermediate and regeneration of the NHC catalyst results in functionalization of the carbonyl group. Thioesters with a β-chiral center were prepared in a redox-neutral transformation with an excellent yield and ee.
42. Visible Light Mediated [4+2] Cycloaddition of Styrenes Synthesis of Tetralin Derivatives
Leifeng Wang, Fengjin Wu, Jiean Chen, David A. Nicewicz,* and Yong Huang*, Angew. Chem. Int. Ed. 2017, 56, 6896-6900.
We report a formal [4+2] cycloaddition reaction of styrenes under visible-light catalysis. Two styrene molecules with different electronic or steric properties were found to react with each other in good yield and excellent chemo- and regioselectivity. This reaction provides direct access to polysubstituted tetralin scaffolds from readily available styrenes. Sophisticated tricyclic and tetracyclic tetralin analogues were prepared in high yield and up to 20/1 diasteroselectivity from cyclic substrates.

Fei Wang, Jiean Chen* and Yong Huang*, Synlett 2017,28, 1300-1304. (Invited Synlett Cluster)
An enantioselective [3+2] cyclization is reported for the construction of a chiral oxazoline skeleton in moderate yield and up to 97% ee. The reactivity and stereochemical discrimination originate from the noncovalent interaction and orientation of a bifunctional catalyst. The novel combination of an α-keto ester and an α-isocyanoacetate establishes an oxazoline which could be a potential chiral ligand for metalmediated catalysis, and also could be easily converted into an optically active β-hydroxy-α-amino acid.
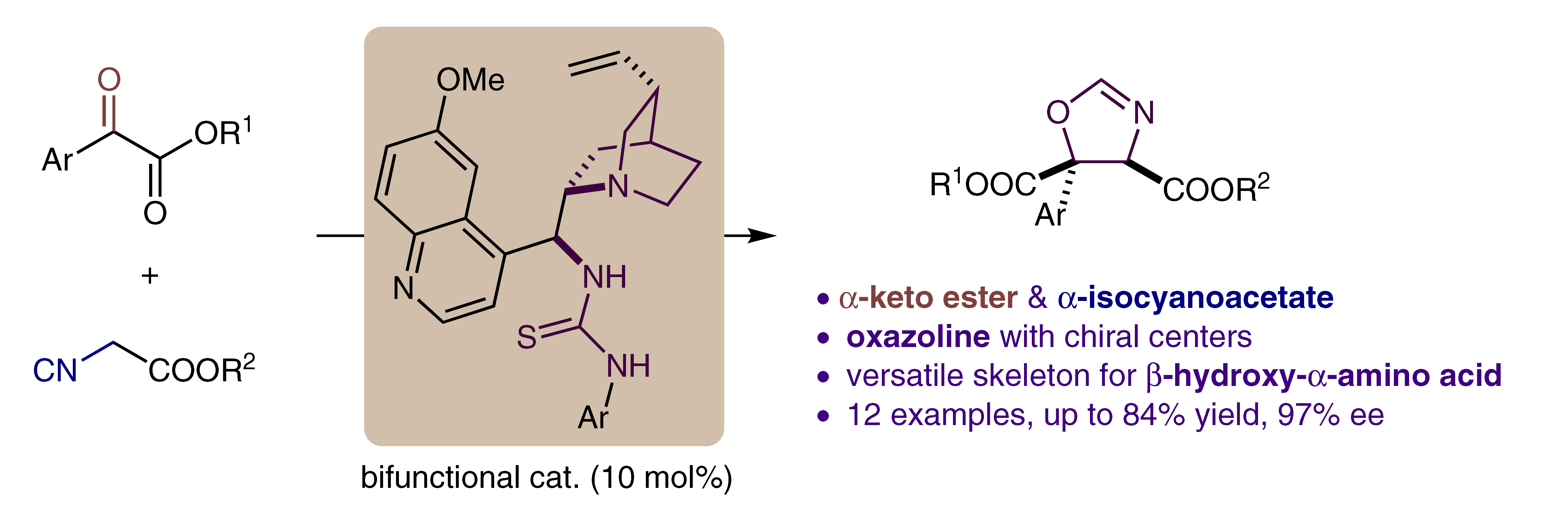
40. A Migratory Ether Formation Route to Medium-Sized Sugar Mimetics
Hao Jiang, Li-Ping Xu, Yan Fang, Zhen-Xing Zhang, Zhen Yang,* and Yong Huang*, Angew. Chem. Int. Ed., 2016, 55, 14340 –14344.
Polyol-substituted cyclic ethers are fundamental building blocks of biomolecules. The position and stereochemistry of multiple hydroxy substituents of cyclic ethers play a central role in their biological function. Current methods for the synthesis of such structures are limited to “naked” ring products with no or few substituents. Here we describe a general route to medium-sized polyol cyclic ethers using a migratory ether formation strategy. In contrast to the common pathway of direct opening of epoxides, Me3Al was found to promote an unprecedented ether addition reaction, opening a neighboring epoxide. The resulting oxonium intermediate triggers a 1,3-methyl shift to yield 2-deoxyribital products. When the hemiacetal auxiliary is a monosaccharide, the sugar ring is expanded by four atoms to give the corresponding 9- to 11-membered analogues. This method provides an entry into the untapped chemical space of medium-sized sugar mimetics.
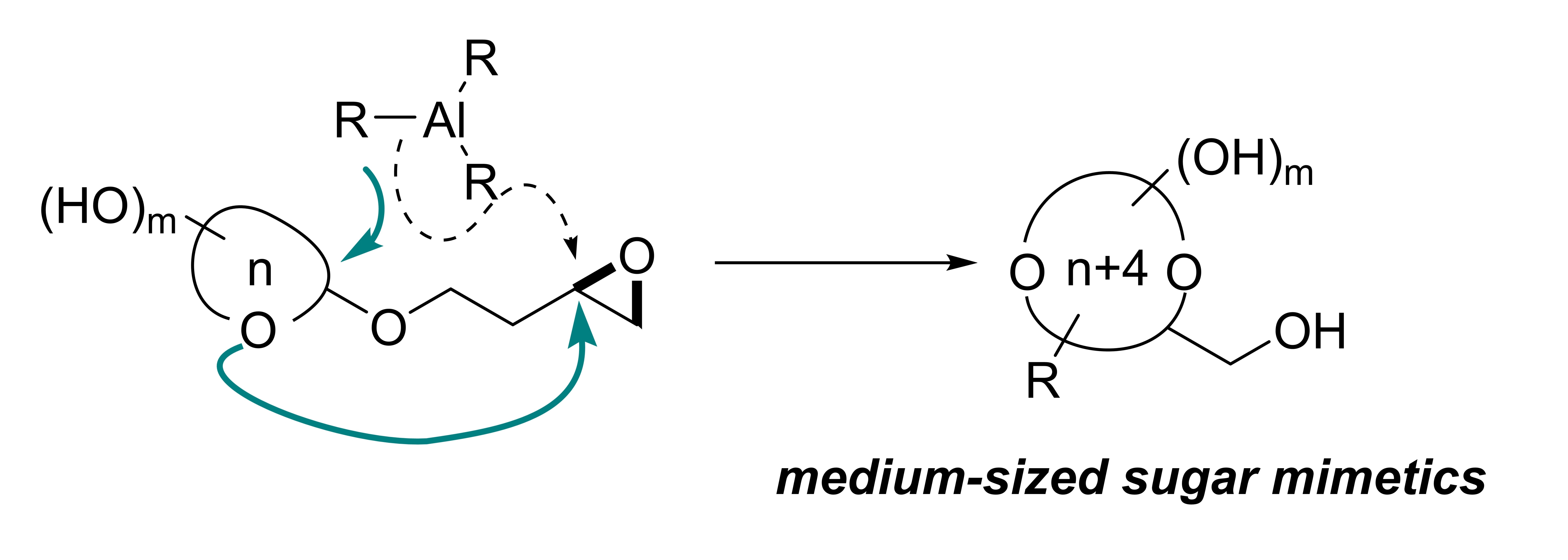
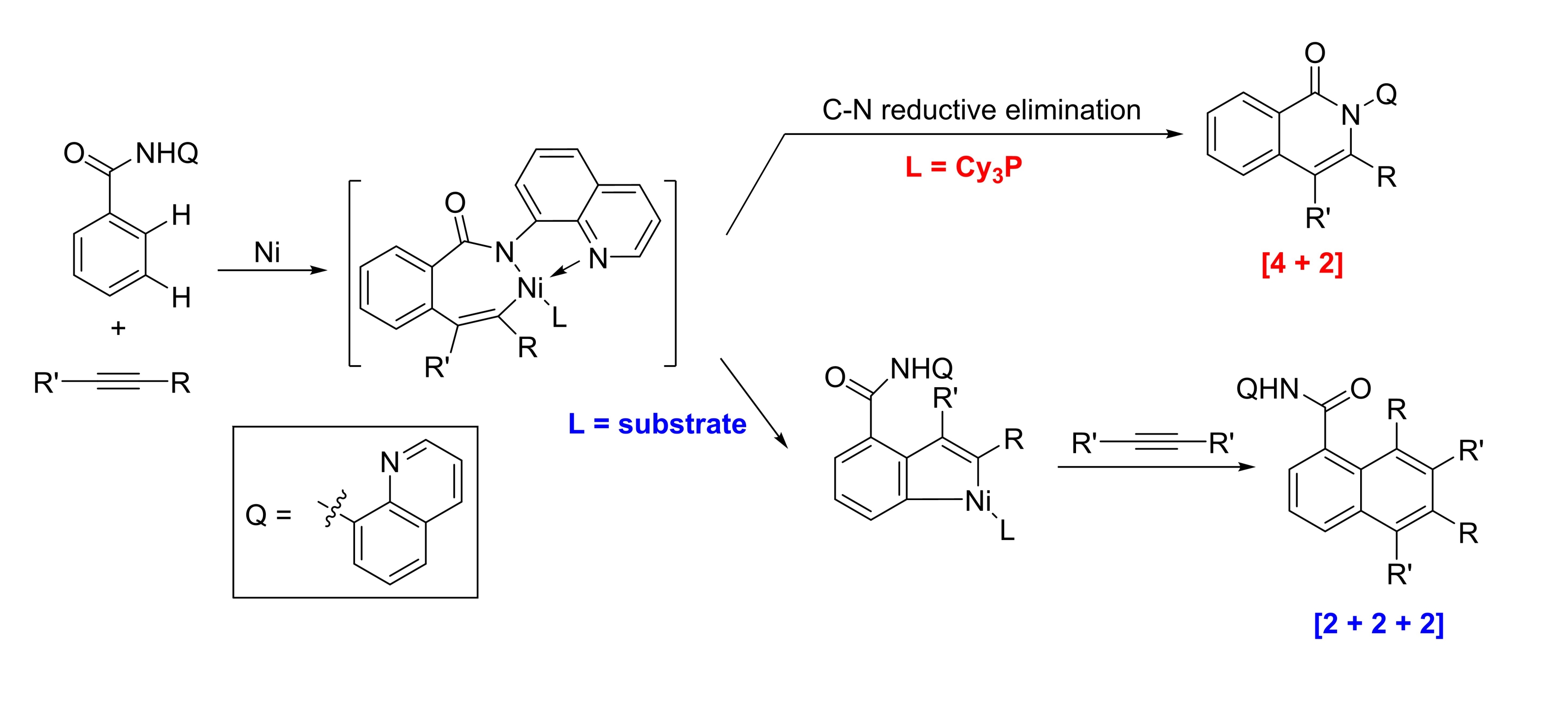
39. Diverting C−H Annulation Pathways Nickel-Catalyzed Dehydrogenative Homologation of Aromatic Amides
Zhiqi He and Yong Huang*, ACS Catal. 2016, 6, 7814−7823.
Direct homologation of aromatic amides with internal alkynes has been accomplished via a nickel-catalyzed sequential C−H activation reaction. The use of a rigid chelating group and a strong aprotic polar solvent successfully divert the classical [4 + 2] annulation to the [2 + 2 + 2] homologation pathway. This transformation is promoted by a simple nickel catalyst without the need of stoichiometric metal oxidants. Mechanistic studies support an unusual substrate-assisted ligand exchange process. NMR and X-ray data suggest a [5,5] Ni-bridged metallacycle as the catalyst resting state. Substrate assisted directing group swap plays an important role for the subsequent meta-C-H insertion. In contrast, [4 + 2] annulation can be accomplished using a bulky, electron-rich phosphine ligand, which favors rapid reductive C−N elimination.
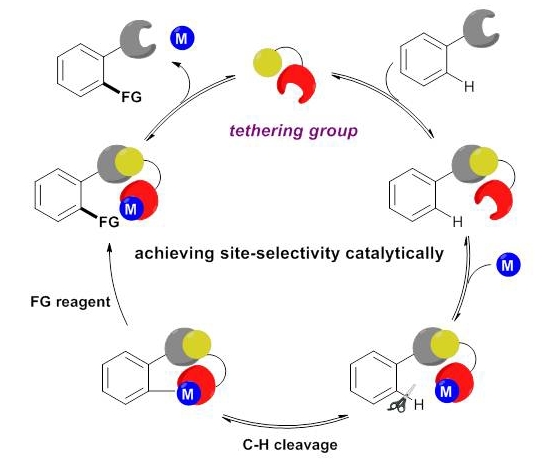
Huan Sun, Nicolas Guimond* and Yong Huang*, Org. Biomol. Chem., 2016,14, 8389–8397.(Invited Perspective)
Transition metal-catalyzed C–H bond insertion is one of the most straightforward strategies to introduce functionalities within a hydrocarbon microenvironment. For the past two decades, selective activation and functionalization of certain inert C–H bonds have been made possible with the help of directing groups (DGs). Despite the enormous advances in the field, an overwhelming majority of systems require two extra steps from their simple precursors: installation and removal of the DGs. Recently, traceless and multitasking groups were invented as a partial solution to DG release. However, installation remains largely unsolved. Ideally, a transient, catalytic DG would circumvent this problem and increase the stepand atom-economy of C–H functionalization processes. In this review, we summarize the recent development of the transient tethering strategy for C–H activation reactions.
37. Ligand-Assisted Palladium(II)/(IV) Oxidation for sp3 C-H Fluorination
Huan Sun, Yi Zhang, Ping Chen, Yun-Dong Wu*, Xinhao Zhang* and Yong Huang*, Adv. Synth. Catal., 2016, 358, 1946.(Very Important Paper, Front Cover)
The direct functionalization of inert sp3 C-H bonds is limited to a few bond types. Although the activation of sp3 C-H bonds can be accomplished under mild conditions using palladium catalysts, the subsequent functionalization is not trivial due to the high energy required to convert palladium(II) to palladium(IV). We have systematically studied the palladium oxidation using computation-guided experiments for reactions involving strong chelation control. We find that a mild external ligand could significantly accelerate the oxidation of palladium(II) to palladium(IV) for strong bidentate directing groups.The acceleration is believed to be a result of ligand stabilization of both the palladium(II) and palladium(IV) intermediates.

36. New frontiers of N-heterocyclic carbene catalysis
Jiean Chen and Yong Huang*, Sci. China Chem., 2016, 59, 251(Invited Perspective)
The multidimensional electronic characteristics of NHC offer great opportunities for catalysis innovation. The synthetic potential of many recently discovered new aspects of NHC remains to be further explored. Deep mechanistic understanding of NHC activation will inevitably lead to invention of new synthetic processes, new catalysts with higher efficiency and selectivity, and novel generic modes of catalysis. We expect this field will continue to blossom and remain at the center of synthetic innovation.

Pengfei Yuan, Sixuan Meng, Jiean Chen* and Yong Huang*, Synlett, 2016, 27, 1068.(Invited Synlett Cluster)
We report an asymmetric sulfa-Michael reaction of α,β-unsaturated amides and esters using a chiral N-heterocyclic carbene as the HOMO-raising organocatalyst. We discovered an interesting correlation between 13C NMR shifts of substrates and ee of their products. More electron-deficient Michael acceptors afforded higher enantioselectivity.
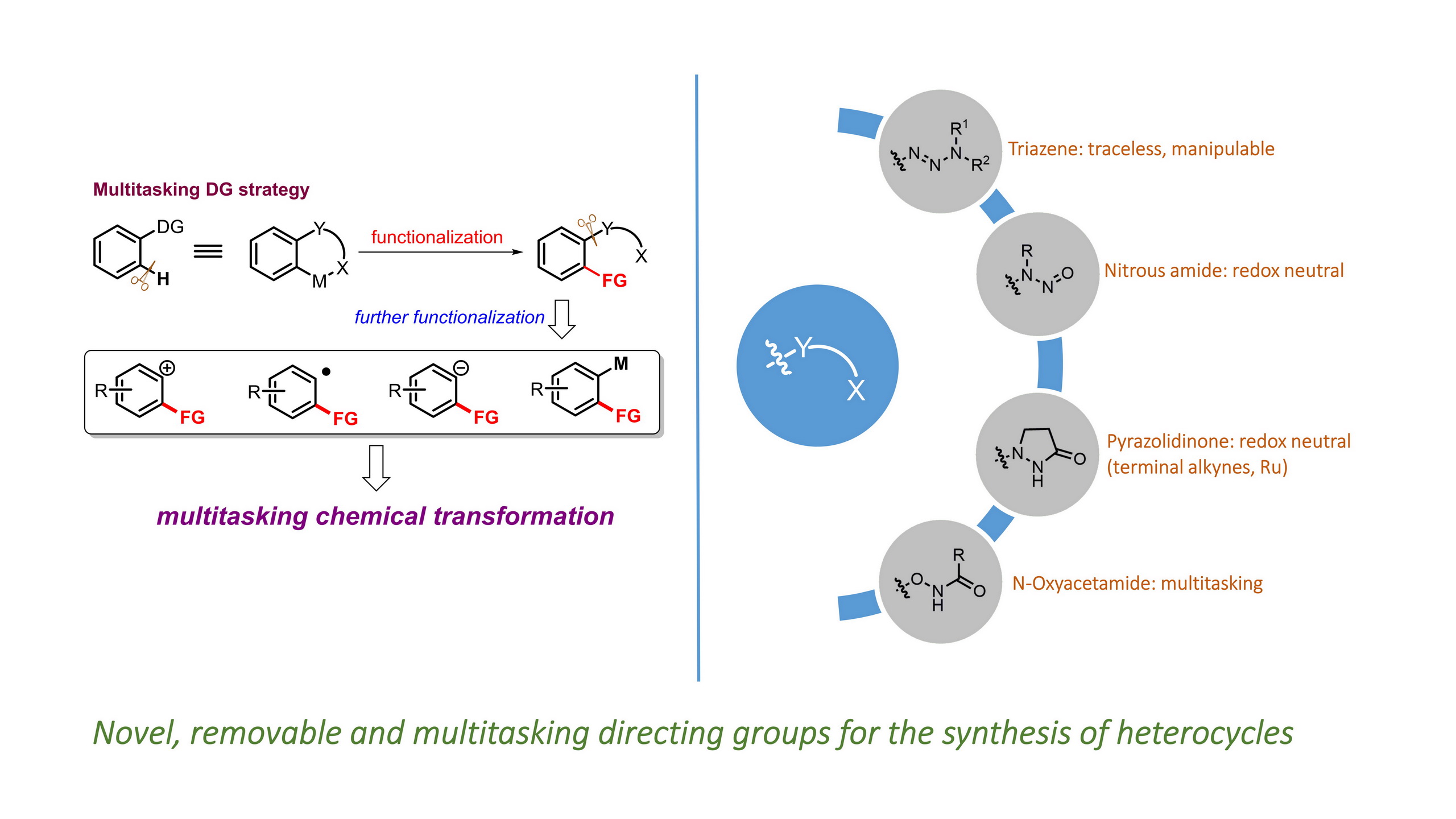
Huan Sun and Yong Huang*, Synlett. 2015, 26, 2751. (Invited Account)
Selective carbon–hydrogen activation reactions can be accomplished in a predictive manner using directing auxiliaries. However, the majority of directing groups discovered to date are difficult to remove or to transform into a desirable functionality. Recently, removable, cleavable, and redox-neutral directing groups have been developed that significantly broaden both the substrate scope and synthetic diversity of carbon–hydrogen functionalization reactions. In this short account, we summarize recent progress we have made in the development of multitasking (removable, cleavable, redox-neutral, manipulable) directing groups for carbon–hydrogen activation reactions.
Leming Wang, Jiean Chen and Yong Huang*, Angew. Chem. Int. Ed. 2015, 54, 15414.
The aza-Michael addition reaction is a vital transformation for the synthesis of functionalized chiral amines. Despite intensive research, enantioselective aza-Michael reactions with alkyl amines as the nitrogen donor have not been successful. We report the use of chiral N-heterocyclic carbenes (NHCs) as noncovalent organocatalysts to promote a highly selective aza-Michael reaction between primary alkyl amines and β-trifluoromethyl β-aryl nitroolefins. In contrast to classical conjugate-addition reactions, a strategy of HOMO-raising activation was used. Chiral trifluoromethylated amines were synthesized in high yield (up to 99 %) with excellent enantioselectivity (up to 98 % ee).
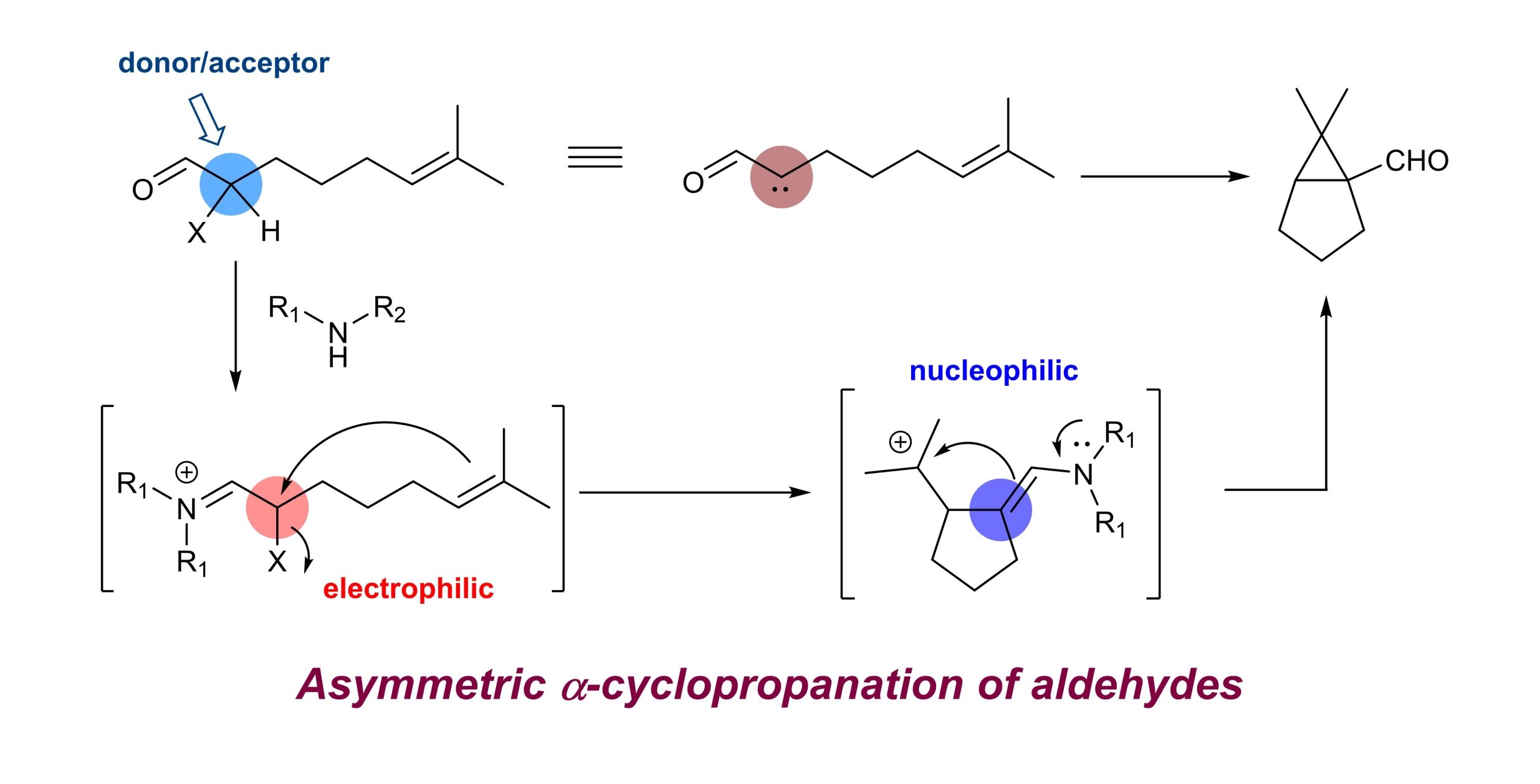
32. Asymmetric intramolecular α-cyclopropanation of aldehydes using a donor/acceptor carbene mimetic
Chaosheng Luo, Zhen Wang and Yong Huang*, Nature Commun. 2015, 6, 10041.
Enantioselective α-alkylation of carbonyl is considered as one of the most important processes for asymmetric synthesis. Common alkylation agents, that is, alkyl halides, are notorious substrates for both Lewis acids and organocatalysts. Recently, olefins emerged as a benign alkylating species via photo/radical mechanisms. However, examples of enantioselective alkylation of aldehydes/ketones are scarce and direct asymmetric dialkylation remains elusive. Here we report an intramolecular α-cyclopropanation reaction of olefinic aldehydes to form chiral cyclopropane aldehydes. We demonstrate that an α-iodo aldehyde can function as a donor/acceptor carbene equivalent, which engages in a formal [2 + 1] annulation with a tethered double bond. Privileged bicyclo[3.1.0]hexane-type scaffolds are prepared in good optical purity using a chiral amine. The synthetic utility of the products is demonstrated by versatile transformations of the bridgehead formyl functionality. We expect the concept of using α-iodo iminium as a donor/acceptor carbene surrogate will find wide applications in chemical reaction development.
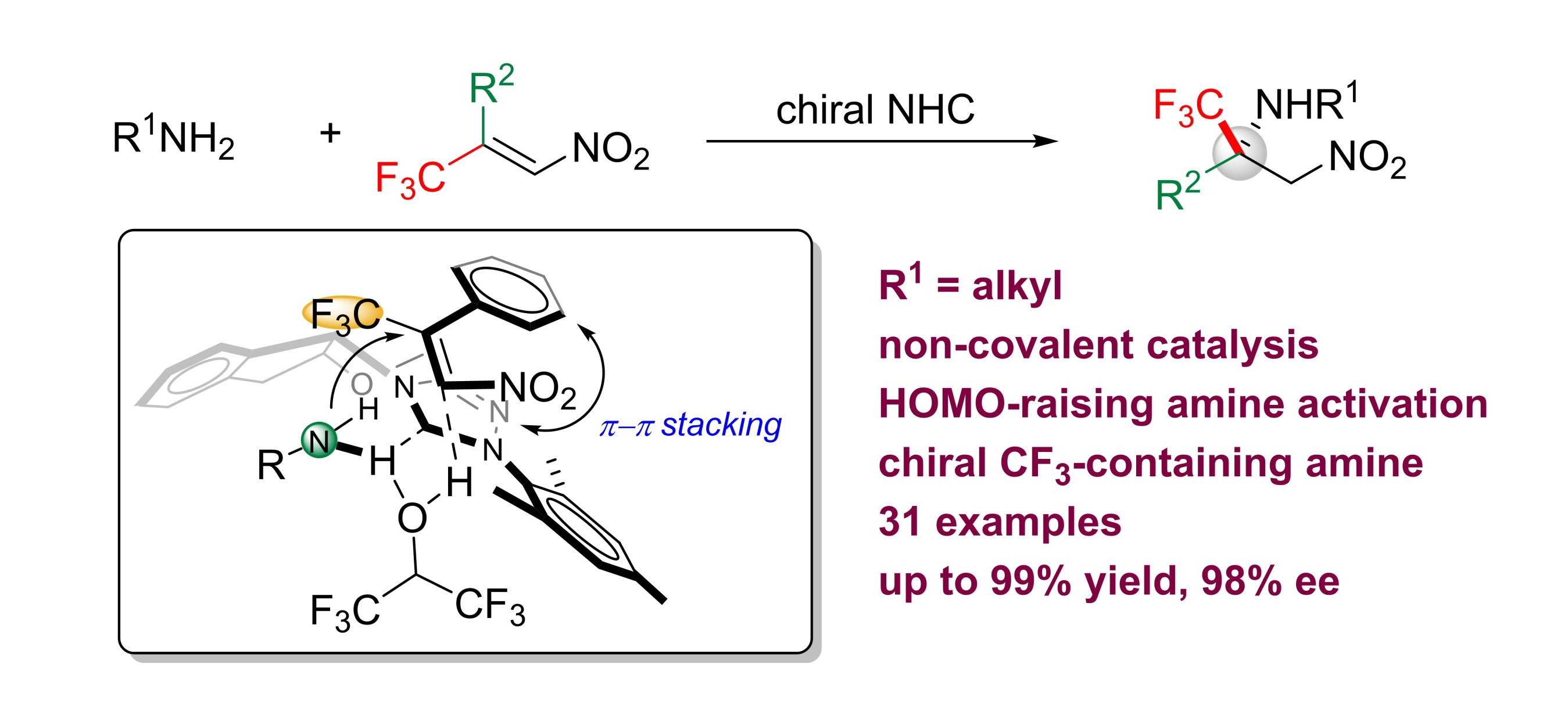
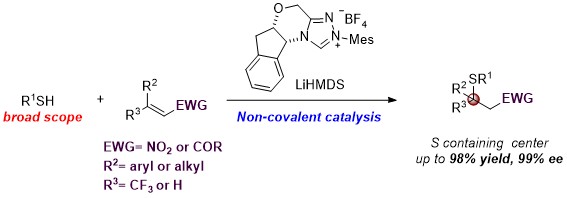
31. Highly enantioselective sulfa-Michael addition reactions using N-heterocyclic carbene as a noncovalent organocatalyst
Jiean Chen, Sixuan Meng, Leming Wang, Hongmei Tang and Yong Huang*, Chem. Sci. 2015, 6, 4184.
We report thefirst asymmetric sulfa-Michael addition (SMA) reactions using a chiralN-heterocyclic carbene(NHC) as a non-covalent organocatalyst. We demonstrate that a triazolium salt derived NHC functions as an excellent Brønsted base to promote enantioselective carbon–sulfur bond formation. The reaction is applicable to a wide range of thiols and electrophilic olefins. Notably, quaternary chiral centers bearing both an S atom and a CF3 group were synthesized with excellent asymmetric control. Mechanistic studies suggest that the facial discrimination is likely to be guided by non-covalent interactions: hydrogen bonding and π–π stacking.
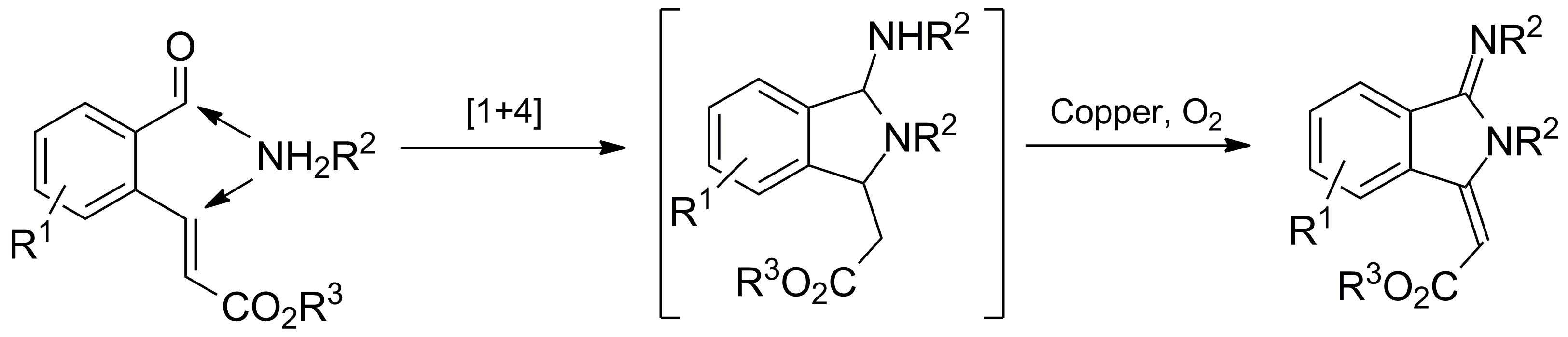
30. A Copper-Catalyzed Aerobic Domino Process for The Synthesis of Isoindolin-1-ylidene Derivatives
Hu Chen, Qian Wang and Yong Huang*, Tetrahedron 2015, 71, 3632. (Invited Contribution for a Special Issue)
A convenient and efficient copper-catalyzed aerobic cascade reaction has been developed for the synthesis of the pharmacologically relevant isoindolin-1-ylidene scaffold. We discovered that various ortho-formyl cinnamates could react smoothly with different amines in the presence of a commercially available copper catalyst under mild aerobic conditions. Isoindolin-1-ylidene derivatives were assembled in one pot in moderate to good yields. This method features amine annulation and double dehydrogenation, representing high atomic efficiency. Its product could be further converted to the privileged isoindolinone pharmacophore.
Xijian Li, Siyu Peng, Li Li and Yong Huang*, Nature Commun. 2015, 6, 6913.
Electron-rich dienes have revolutionized the synthesis of complex compounds since the discovery of the legendary Diels–Alder cycloaddition reaction. This highly efficient bond-forming process has served as a fundamental strategy to assemble many structurally formidable molecules. Amino silyloxy butadienes are arguably the most reactive diene species that are isolable and bottleable. Since the pioneering discovery by Rawal, 1-amino-3-silyloxybutadienes have been found to undergo cycloaddition reactions with unparalleled mildness, leading to significant advances in both asymmetric catalysis and total synthesis of biologically active natural products. In sharp contrast, this class of highly electron-rich conjugated olefins has not been studied in non-cycloaddition reactions. Here we report a simple synthesis of tetrasubstituted 1-silyloxy-3-aminobutadienes, a complementarily substituted Rawal’s diene. This family of molecules is found to undergo a series of intriguing chemical transformations orthogonal to cycloaddition reactions. Structurally diverse polysubstituted ring architectures are established in one step from these dienes.
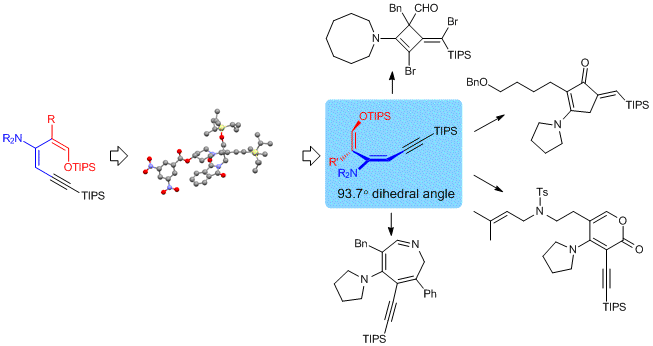
28. Direct Fluorination of Styrenes
Qian Shao, and Yong Huang*, Chem. Commun. 2015, 51, 6584-6586.
We have developed a practical method to synthesize fluorostyrene compounds. A mild and regioselective mono-fluorination reaction occurred smoothly for various di- and trisubstituted styrenes in the presence of RuCl3 and N-fluorobenzenesulfonimide (NFSI). A tandem alkyne hydroarylation–olefin fluorination reaction was also developed using an Au catalyst.
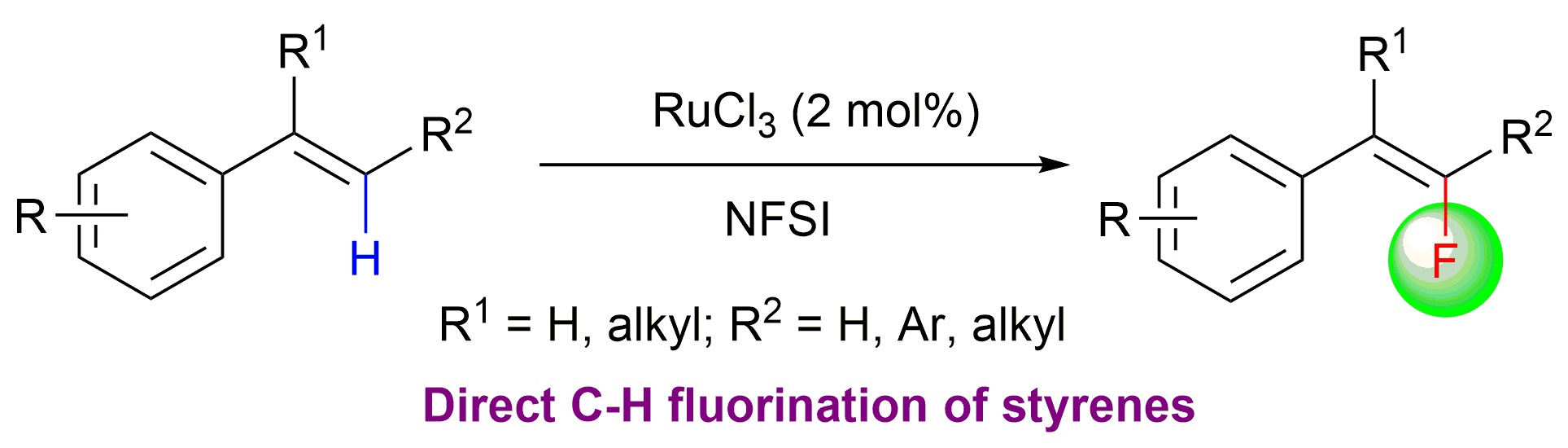
27. Synthesis of Indolo[2,1-a]isoquinolines via a Triazene-Directed C–H Annulation Cascade
Huan Sun, Chengming Wang, Yun-Fang Yang, Ping Chen, Yun-Dong Wu*, Xinhao Zhang* and Yong Huang*, J. Org. Chem. 2014, 79, 11863-11872.
Indole-containing polyaromatic scaffolds are widely found in natural products, pharmaceutical agents, and π-conjugated functional materials. Often, the synthesis of these highly valuable molecules requires a multistep sequence. Therefore, a simple, one-step protocol to access libraries of polyaromatic indole scaffolds is highly desirable. Herein we describe the direct synthesis of polysubstituted indolo[2,1-a]isoquinoline analogues via a double C–H annulation cascade using triazene as an internally cleavable directing group. Evidence from HRMS and theoretical calculations suggests that an unprecedented 1,2-alkyl migration might be responsible for the in situ cleavage of the directing group. Both kinetic isotope effects and DFT calculations suggested that the alkyne insertion step is rate-limiting for the second C,N annulation reaction.
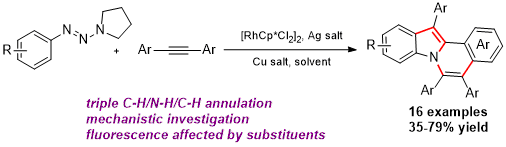
Zhenxing Zhang, Hao Jiang and Yong Huang*, Org. Lett. 2014, 16, 5976–5979.
The first Ru-catalyzed redox-neutral C–H activation reaction via N–N bond cleavage is reported. Pyrazolidin-3-one is demonstrated as an internally oxidative directing group that enables C–H annulation reactions with a broad scope of alkynes, including previously incompetent terminal alkynes. Pharmacologically privileged 3-(1H-indol-1-yl)propanamides were synthesized in high yields.
25. Directed Arenealkyne Annulation Reactions via Aerobic Copper Catalysis
Yi Zhang, Qian Wang, Huidong Yu* and Yong Huang*, Org. Biomol. Chem. 2014, 12, 8844-8850.
We describe a straightforward protocol for a smooth dehydrogenative annulation reaction between various arenes and terminal alkynes using a catalytic amount of CuBr2 and molecular oxygen. 3-Methyleneisoindoline derivatives are prepared in high yields.
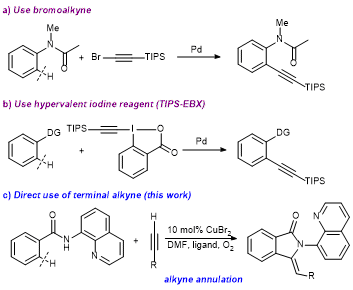
24. Highly ortho-Selective Trifluoromethylthiolation Reactions using a Ligand Exchange Strategy
Weiyu Yin, Zhaofeng Wang and Yong Huang*, Adv. Synth. Catal. 2014, 356, 2998 – 3006.
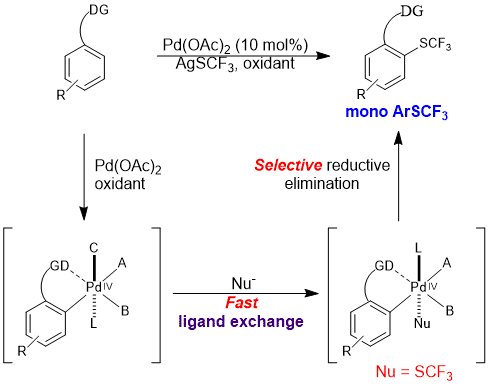
The trifluoromethylthio group (![[BOND]](http://onlinelibrarystatic.wiley.com/undisplayable_characters/00f8ff.gif) SCF3) is a highly privileged modifier for drug molecules. Direct CH trifluoromethylthiolation reactions are highly valuable synthetic tools for the late-stage modification of drug candidates, which have so far been underexplored. We report a palladium-catalyzed ortho-selective mono-trifluoromethylthiolation reaction of arenes. The reaction proceeds through a key ligand exchange pathway using readily available silver trifluoromethylthiolate (AgSCF3). Acetic acid was found to be crucial to minimize oxidative dimerization of the starting materials and to facilitate the “SCF3−” transfer. We expect this strategy to have broad applications in CH functionalization reactions using a stand-alone oxidant and various anions.
SCF3) is a highly privileged modifier for drug molecules. Direct CH trifluoromethylthiolation reactions are highly valuable synthetic tools for the late-stage modification of drug candidates, which have so far been underexplored. We report a palladium-catalyzed ortho-selective mono-trifluoromethylthiolation reaction of arenes. The reaction proceeds through a key ligand exchange pathway using readily available silver trifluoromethylthiolate (AgSCF3). Acetic acid was found to be crucial to minimize oxidative dimerization of the starting materials and to facilitate the “SCF3−” transfer. We expect this strategy to have broad applications in CH functionalization reactions using a stand-alone oxidant and various anions.
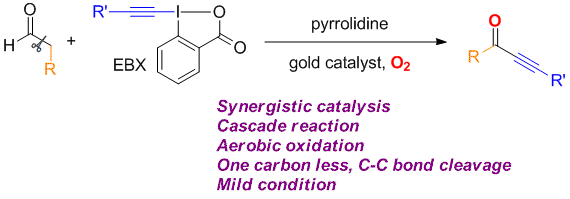
Zhaofeng Wang, Li Li and Yong Huang*, J. Am. Chem. Soc. 2014, 136, 12233–12236.
We describe a direct synthesis of various ynones from readily available aldehydes and hypervalent alkynyl iodides. In this method, a gold catalyst and a secondary amine work synergistically to produce the trisubstituted allenyl aldehyde, which can be converted to the desired ynone through an in situ C–C bond oxidative cleavage using molecular oxygen.
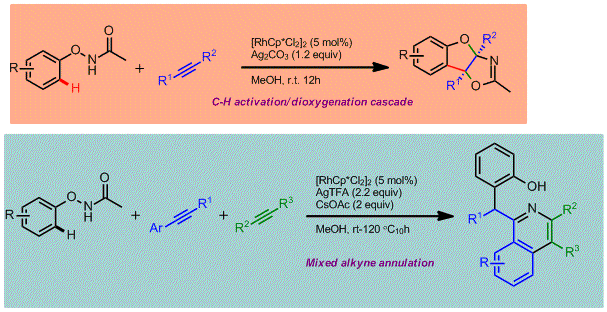
Ying Chen, Dongqi Wang, Pingping Duan, Rong Ben, Lu Dai, Xiaoru Shao, Mei Hong, Jing Zhao* and Yong Huang*, Nature Commun. 2014, 5, 4610.
The C–H activation strategy has become one of the preferred methods to introduce chemical functionality to a chemically inert carbon atom. Intensive efforts have been devoted to developing either versatile bond formations (product structural diversity) or effective directing groups (substrate site selectivity). From the views of medicinal and synthetic practitioners, the C–H activation approach remains inadequate due to its limitation to point-to-point derivatization. Direct assembly of 3D molecular complexity in a single step remains elusive for this strategy. Towards this goal, a multitasking functional group is required to accomplish several missions in one pot: site selecitivity, cleavability and redox versatility. We demonstrate that an oxyacetamide group is such a multifunctional warhead that enables a series of C–H functionalization cascades and allows direct access to structurally diverse polycyclic heterocyles in one pot. The progress of these reaction cascades were fully controlled by oxidants and temperature. The proliferation of the reaction chain can be extended to a four-step cascade.
21. Asymmetric catalysis with N-heterocyclic carbenes as non covalent chiral templates
Jiean Chen and Yong Huang*, Nature Commun. 2014, 5, 3437.
N-heterocyclic carbenes are a class of persistent carbenes stabilized by adjacent heteroatoms that are part of a heterocycle. They play a central role in multiple enzymatic biosynthetic reactions that involve thiamine diphosphate. Inspired by this biocatalysis machinery, N-heterocyclic carbenes have emerged as one of the most versatile classes of organocatalysts for organic reactions. However, the asymmetric synthesis of carbon–carbon bonds through a non-covalent interaction mechanism has not been previously established for chiral carbenes. Here, we report an N-heterocylic carbene-catalysed, highly enantioselective process that uses weak hydrogen bonds to relay asymmetric bias. We find that catalytic amounts of hexafluoroisopropanol are the critical proton shuttle that facilitates hydrogen transfer to provide high-reaction rates and high enantioselectivity. We demonstrate that a successful asymmetric reaction of this type can be accomplished through a rational design that balances the pKa values of the substrate, the carbene precursor and the product.
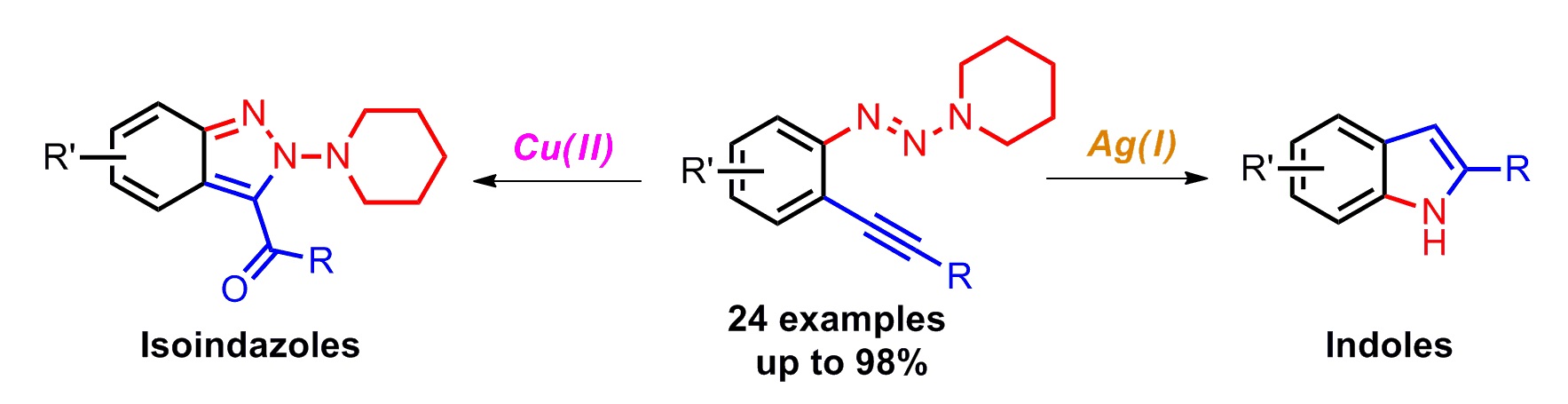
Yan Fang, Chengming Wang, Shengqin Su, Haizhu Yu* and Yong Huang*, Org. Biomol. Chem. 2014,12, 1061-1071.
We described two orthogonal heterocycle syntheses, where an arene bearing both an alkyne and a triazene functionality underwent two distinct cyclization pathways mediated by different transition metals. Starting from the same substrates, a synthesis of 2H-indazole was accomplished by a Cu(II) salt promoted oxidative cyclization, while 2-substituted indoles could be accessed via a Ag(I) salt mediated N–N bond cleavage. This method represents the first synthesis of indoles from alkynyl triazenes. Computational analysis was performed for both reaction pathways, supporting a Lewis acid role for Cu and a π-acid catalysis for Ag.

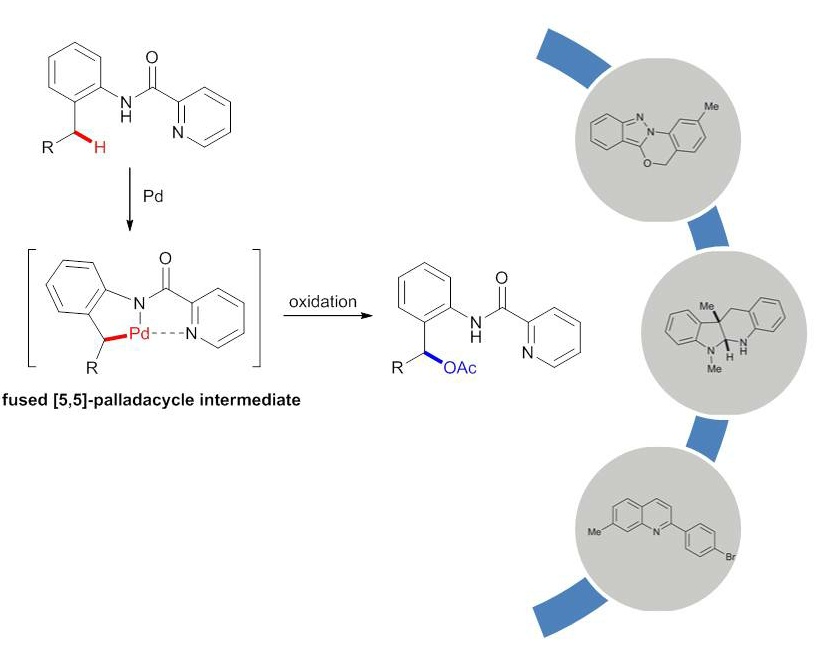
Tao Cheng, Weiyu Yin, Yi Zhang, Yingnan Zhang and Yong Huang*, Org. Biomol. Chem. 2014,12, 1405-1411.
A general palladium catalyzed acetoxylation of benzylic C–H bonds has been developed. Picolinamides serve as an excellent directing group for the C–H activation of benzylic methyls. A wide range of 2-amino benzyl alcohol analogues were synthesized in good yields. The products demonstrated broad synthetic utilities toward various benzo-fused heterocycles. Mechanistic studies revealed the key rate-limiting C–H insertion step, which could be affected by the substitution pattern of the parent arene.
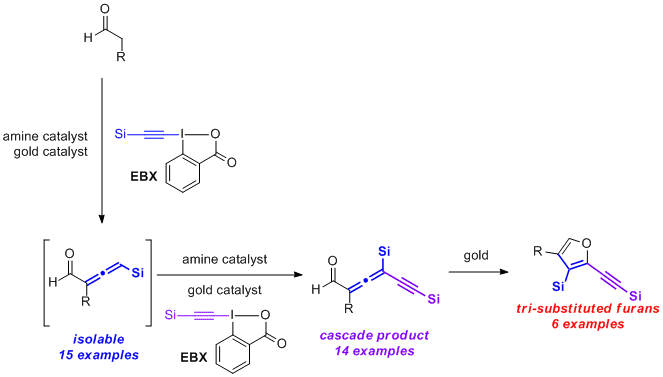
Zhaofeng Wang, Xijian Li, Yong Huang*, Angew. Chem. Int. Ed. 2013, 42, 14219-14223.
Carbonyl-substituted allenes are highly important synthetic intermediates for a number of heterocycles and strained-ring systems. However, chemistry of allenyl aldehydes has not been explored as extensively as their ketone, ester, or amide analogues because of a lack of general synthetic methods. Described herein is the first direct α-vinylidenation of aldehydes and an α-vinylidenation/γ-functionalization cascade to access tri- and tetrasubstituted allenyl aldehydes using a combination of a gold catalyst and an secondary amine. The reactive enamine intermediate of an aldehyde reacts with the gold-activated hypervalent silylethynyl benziodoxolone to selectively generate the corresponding trisubstituted allenyl aldehyde. The allenyl aldehyde can further react with another equivalent of the alkynylation reagent or other electrophiles to afford tetrasubstituted allenes bearing an aldehyde group, an acetylene, and a halogen functionality. This method enables rapid access to polysubstituted furans from aldehydes.

Junlin Zhang, Leming Wang, Qi Liu, Zhen Yang*, and Yong Huang*, Chem. Commun. 2013, 49, 11662-11664.
α,β-Unsaturated ketones and aldehydes have been synthesized from their corresponding silyl enol ethers in a straightforward protocol involving a visible-light promoted organocatalytic, aerobic oxidation reaction. A cheap organic dye was used catalytically in these reactions as the photosensitizer.

Qian Shao, Jiean Chen, Meihua Tu, David W. Piotrowski and Yong Huang*, Chem. Commun. 2013, 49, 11098-11100.
An enantioselective organocatalytic process for the one-step synthesis of poly-substituted 1,2,4-triazolines is reported. The heterocycle formation is believed to go through a step-wise mechanism of nucleophilic addition of an azlactone to an azodicarboxylate in the presence of an organic base catalyst, followed by a TMSCHN2 mediated heterocyclization. Both theoretical calculations and experimental evidence suggest the pre-organization of the transition state for the chirality determining step via a unique 7-membered intramolecular hydrogen bonding.
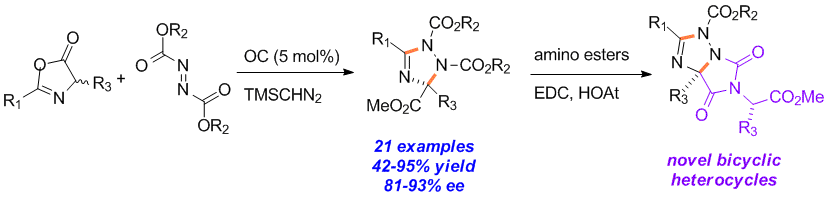
15. Traceless Directing Strategy Efficient Synthesis of N-Alkyl Indoles via Redox-Neutral C–H Activation
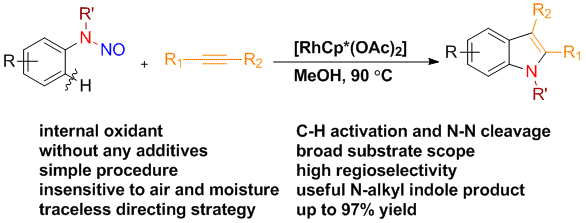
Chengming Wang and Yong Huang*, Org. Lett. 2013, 15, 5294-5297.
A general protocol for the synthesis of N-alkyl indoles has been developed via a redox neutral C–H activation strategy using a traceless nitroso directing group. A broad scope of substituted N-alkyl indoles has been prepared in good to excellent yields using a very simple Rh catalyst system in the absence of an external oxidant or any other additive. Good to excellent regioselectivity has been achieved for asymmetrically disubstituted acetylenes.
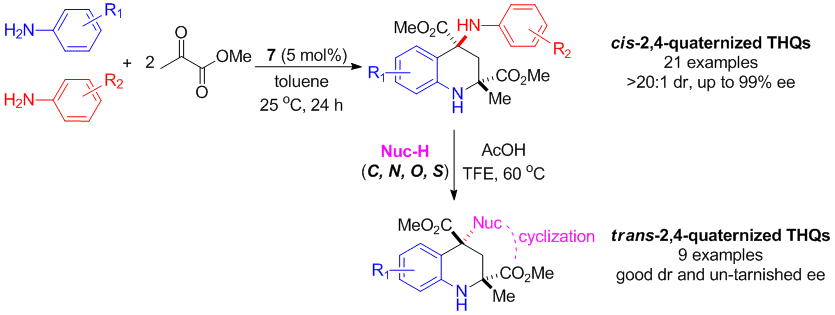
Chaosheng Luo and Yong Huang*, J. Am. Chem. Soc. 2013, 135, 8193–8196.
Highlighted by SYNFACTS, 2013, 9(8), 0898; DOI-10.1055s-0033-1339442.
Tetrahydroquinolines containing two quaternary stereogenic centers were synthesized with excellent ee and dr via a four-component cyclization reaction catalyzed by a chiral phosphoric acid. High chemoselectivity was achieved by differentiating anilines with similar reactivities to yield diverse “hybrid” products. The chirality of the quaternary C4 atom of the 4-aminotetrahydroquinoline products was found to undergo highly stereoselective inversion, enabling facile functionalization using a wide range of nucleophiles (C, O, N, and S).
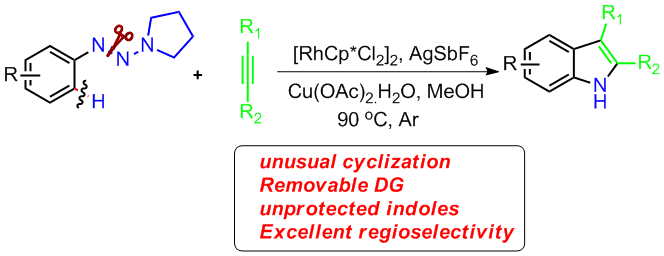
13. General and Efficient Synthesis of Indoles through Triazene Directed C–H Annulation
Chengming Wang, Huan Sun, Yan Fang, and Yong Huang*, Angew. Chem. Int. Ed. 2013, 52, 5795-5798. (cover)
Highlighted by Chinese-Journal-of-Organic-Chemistry-2013-337-1593.
Unprotected indoles are prepared with the title method, which has a wide scope of alkynes. Excellent regioselectivity was accomplished for aryl–alkyl and alkyl–alkyl disubstituted acetylenes. This reaction features an unusual 1,2 rhodium migration and ring-contraction-triggered NN bond cleavage. It allows rapid conversion of the reaction products to several functional molecules.

Tao Cheng, Sixuan Meng, and Yong Huang*, Org. Lett. 2013, 15 , 1958–1961.
Highly functionalized pyrrolidine and piperidine analogues, with up to three stereogenic centers, were synthesized in good yield (50–95%), excellent dr (single isomer), and high ee (>90%) using a Cinchona alkaloid-derived carbamate organocatalyst. High stereoselective synergy was achieved by combining a reversible aza-Henry reaction with a dynamic kinetic resolution (DKR)-driven aza-Michael cyclization. Whereas both reactions proceed with moderate enantioselectivities (50–60% for each step), high enantioselectivities are obtained for the overall products devoid of dr sacrifice.
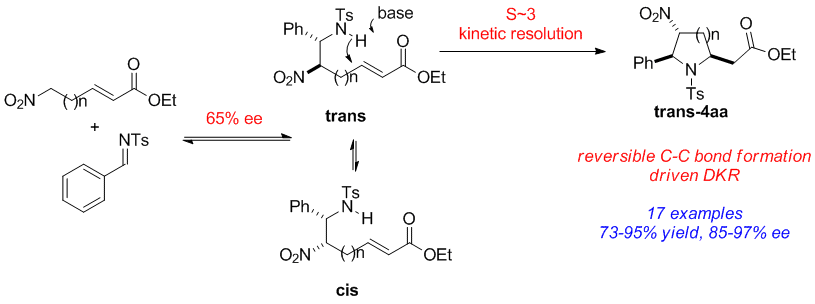
Weiyu Yin, Chengming Wang and Yong Huang*, Org. Lett. 2013, 15, 1850–1853.
Highlighted-by-SYNFACTS-2013-97-0722.-DOI_-10.1055_s-0033-1339227., Highlighted by organic chemistry portal
A mild, aerobic, catalytic process for obtaining nitriles directly from alcohols and aqueous ammonia is described. The reaction proceeds via a dehydrogenation cascade mediated by catalytic CuI, bpy, and TEMPO in the presence of O2. The substrate scope is broad including various functionalized aromatic and aliphatic alcohols. This protocol enabled the one-pot synthesis of various biaryl heterocycles directly from commercially available alcohols.

Hu Chen, Zhaofeng Wang, Yingnan Zhang and Yong Huang*, J. Org. Chem. 2013, 78, 3503-3509.
Highlighted by SYNFACTS, 2013, 9(6), 0602. DOI: 10.1055/s-0033-1338792.
A triple cascade was developed using a simple copper catalyst to trans-selectively access bicyclic isoxazolidines in a one-pot synthesis. This strategy features the in situ generation of nitrones and subsequent trapping by [3 + 2] cycloaddition. In this method, copper serves three catalytic functions: as a Lewis acid for the ene reaction, as an organometallic for aerobic oxidation, and as a Lewis acid for an endo-selective [3 + 2] cycloaddition. The successful merging of aerobic oxidation and Lewis acid catalysis demonstrated efficient cascade synergy.
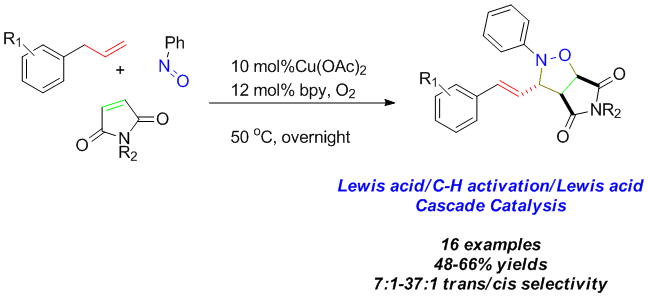
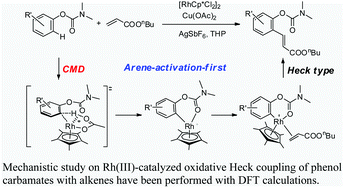
Qi Zhang, Hai-Zhu Yu, Yi-Tong Li, Lei Liu, Yong Huang* and Yao Fu*, Dalton Trans. 2013, 42, 4175-4184.
A systematic theoretical study on the Rh-catalyzed oxidative Heck-coupling of phenol carbamates with alkenes is carried out. Two possible mechanisms (i.e. arene activation-first and alkene activation-first mechanisms) are examined. As to the C–H activation step, four mechanisms including oxidative addition, electrophilic substitution, concerted metallation-deprotonation (CMD), and σ-bond metathesis are evaluated. The calculation results indicate that the arene activation-first mechanism is more favorable for the overall catalytic cycle. This mechanism involves three steps: arene C–H activation at the position ortho to the carbamate directing group affording a six-membered rhodiacycle intermediate, insertion of the alkene double bond into the Rh(III)–aryl bond, and a final β-H elimination step to release the product and re-generate the catalyst. The rate determining step of the overall catalytic cycle is the arene C–H activation step, which is found to proceed through the acetate-assisted CMD mechanism.
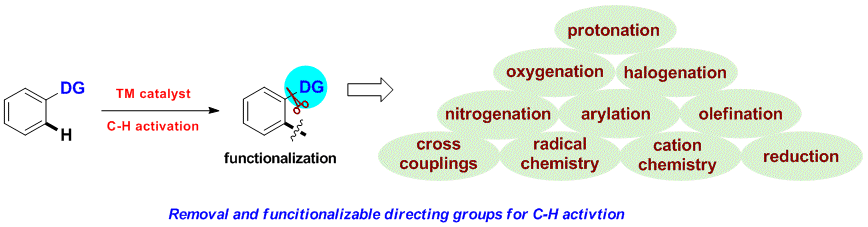
8. Expanding Structural Diversity Removable and Manipulable Directing Groups for C-H Activation
Chengming Wang, Yong Huang*, Synlett. 2013, 24,145.(invited)
The need to use a static directing group represents a major limitation for the emerging field of ortho-selective C-H activation and functionalization. Chemistry was recently developed that allowed partial removal or minor transformations for certain directing groups. Our recent work demonstrated that triazenes were a class of excellent directing groups for C-H activation/functionalization, and subsequent chemical manipulations generated key compounds with synthetic versatility.
Yong Huang, Weiming Gao, Torbjõrn Åkermark, Mingrun Li, BjÖrn Åkermark*, Eur. J. Inorg. Chem. 2012, 27, 4259-4263.
A Fe3S4 complex bridged by azapropanedithiolate (adt), complex 6, was prepared as a potential model of the HOXair state of [FeFe]-hydrogenases. Complex 6 was characterized by IR and 1H NMR spectroscopy, and its structure was deter-mined by X-ray crystallography. The electrochemical studies show that complex 6 is redox-active under acidic conditions, which provides insight into the catalytic mechanism. Hydrogen evolution, driven by visible light, was observed in CH3CN/D2O solution by online mass spectroscopy.
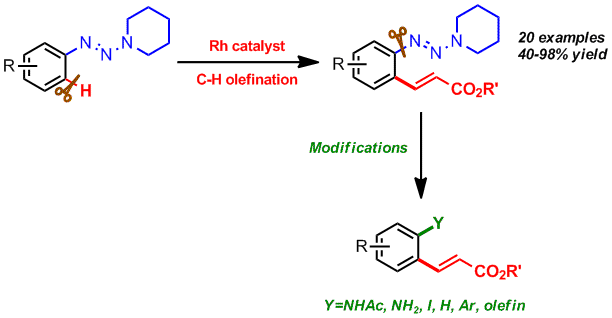
Chengming Wang, Hu Chen, Zhaofeng Wang, Jiean Chen and Yong Huang*, Angew. Chem. Int. Ed. 2012, 51, 7242-7245. (cover)
Highlighted by organic chemistry portal
Diverse opportunities: A Rhodium(III)-catalyzed ortho-selective olefination of arenes using a novel triazene as a directing group is reported. This method exhibits substantial post-functionalization synthetic versatility, overcoming a vital limitation in sp2C-H activation/functionalization products: restrictedstructural diversity.

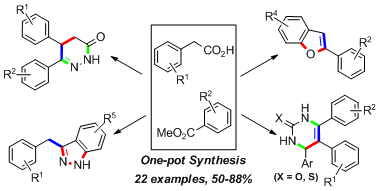
5.One pot synthesis of useful heterocycles in medicinal chemistry using a cascade strategy
Guiyong Wu, Weiyu Yin, Hong C. Shen,* and Yong Huang*, Green. Chem. 2012, 14, 580-585. (cover)
To access useful heterocycles in medicinal chemistry such as pyridazinones, dihydropyrimidinones, and dihydropyrimidinthiones, a “green” mild and highly efficient one-pot triple cascade was developed involving a Claisen–decarboxylation, electrophilic reaction, and subsequent heterocyclization. In addition, indazoles and benzofurans could also be constructed via a double cascade. To develop the cascade process, a direct Claisen–decarboxylation reaction was firstly optimized. This reaction can then couple with electrophilic reactions including alkylation, Michael addition or aldol reaction to enable the preparation of various aryl ketones in a one-pot fashion.

4.General palladium catalyzed aerobic dehydrogenation to generate double bonds
Weiming Gao, Zhiqi He, Yong Qian, Jing Zhao* and Yong Huang*, Chem. Sci. 2012, 3, 883-886.
We describe a general dehydrogenation procedure to form α,β-unsaturated aldehydes, ketones, esters and azobenzenes under very mild conditions, requiring catalytic commercial Pd(OAc)2, a catalytic weak inorganic base and air as the sole oxidant. In the presence of a diazafluorenone ligand, this process converts aliphatic aldehydes to α,β-unsaturated aldehydes in an open-flask fashion at ambient pressure and temperature. A broad spectrum of substrates, including aldehydes, ketones, esters, alcohols and hydrazines, were conveniently dehydrogenated under a relatively uniformed protocol. A mechanism involving β-elimination-driven enolization equilibrium shift was proposed.

Ying-Xiang Gao, Le Chang, Hang Shi, Bo Liang, Kittiya Wongkhan, Duangduan Chaiyaveij, Andrei.S. Batsanov, Todd.B. Marder,* Chuang Chuang Li,* Zhen Yang,* Yong Huang* Adv, Synth. Catal. 2010, 352, 1955-1966.
We report herein the synthesis of novel chiral S,N-heterobidentate thiourea-oxazoline ligands and their application to palladium-catalyzed enantioselective bis(methoxycarbonylation)s of terminal olefins under mild conditions. Copper salts were found to play multiple roles in this reaction. Substituted 2-phenylsuccinates were obtained in >90% yield and up to 84% ee under optimized conditions.
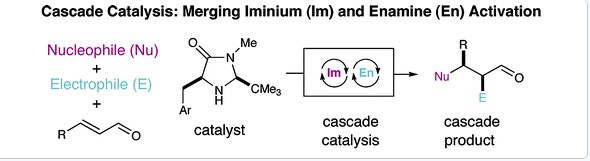
2. Enantioselective Organo-Cascade Catalysis
Yong Huang, Abbas M. Walji , Catharine H. Larsen, and David W. C. MacMillan *, J. Am. Chem. Soc. 2005, 127, 15051-15053.
A new strategy for organocatalysis based on the biochemical blueprints of biosynthesis has enabled a new laboratory approach to cascade catalysis. Imidazolidinone-based catalytic cycles, involving iminium and enamine activation, have been successfully combined to allow a large diversity of nucleophiles (furans, thiophenes, indoles, butenolides, hydride sources, tertiary amino lactone equivalents) and electrophiles (fluorinating and chlorinating reagents) to undergo sequential addition with a wide array of α,β-unsaturated aldehydes. These new cascade catalysis protocols allow the invention of enantioselective transformations that were previously unknown, including the asymmetric catalytic addition of the elements of HF across a trisubstituted olefin. Importantly, these domino catalysis protocols can be mediated by a single imidazolidinone catalyst or using cycle-specific amine catalysts. In the latter case, cascade catalysis pathways can be readily modulated to provide a required diastereo- and enantioselective outcome via the judicious selection of the enantiomeric series of the amine catalysts. A central benefit of combining multiple asymmetric organocatalytic events into one sequence is the intrinsic requirement for enantioenrichment in the second induction cycle, as demonstrated by the enantioselectivities obtained throughout this study (≥99% ee in all cases).
1. Hydrogen bonding: Single enantiomers from a chiral-alcohol catalyst
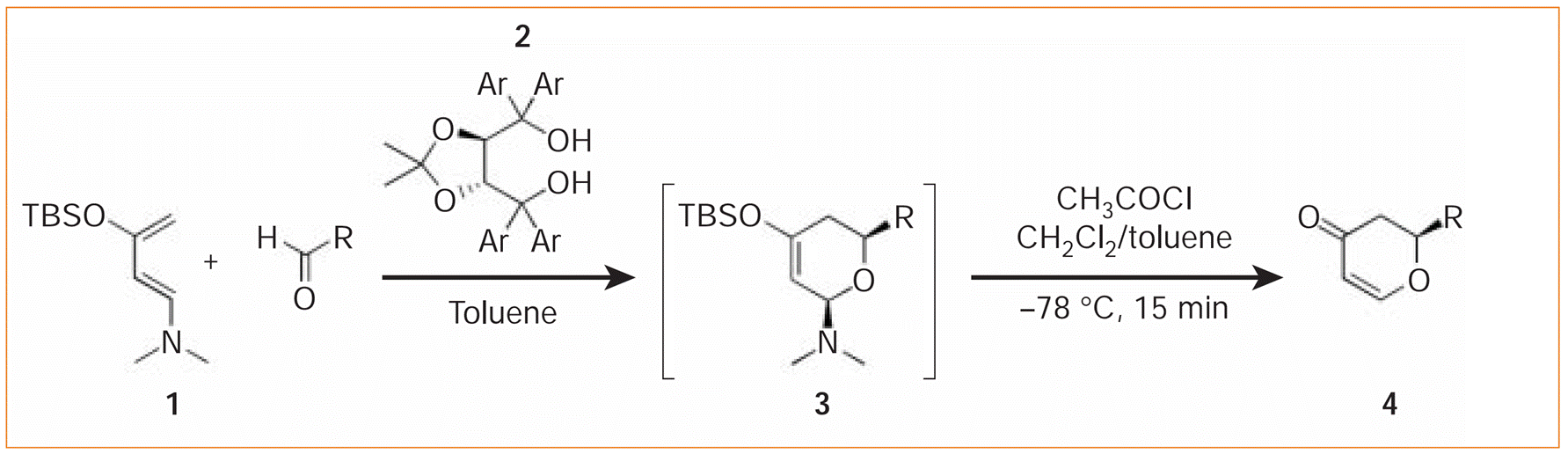
Yong Huang, Aditya K. Unni, Avinash N. Thadani and Viresh H. Rawal*, Nature 2003, 424, 146-146.
Hydrogen bonding acts as a ubiquitous glue to sustain the intricate architecture and functionality of proteins, nucleic acids and many supramolecular assemblies,but this weak interaction is seldom used as a force for promoting chemical reactions. Here we show that a simple chiral alcohol uses hydrogen bonding to catalyse an important family of cycloaddition reactions of a diene with various aldehydes — moreover, this reaction is highly enantioselective, generating only one of the mirror-image forms of each dihydropyran product. This type of catalysis mimics the action of enzymes and antibodies, and is unlike traditional, metal-based catalysts used in organic chemistry.
Patents:
1. “A method to Prepare Nitriles” 201210447319.X
2. "Methods of Preparing Ketones and Their Derivatives" 201110252324.0
3. "Preparation of Cyclopropyl Compounds Containing Pyridine and Pyrimidine Rings as GPR119 Receptor Agonists for the Treatment of Type 2 Diabetes and Related Diseases" WO2009129036
4. "Preparation of Bipiperidinyl Compounds as GPR-119 Agonists for Treating and Preventing Diabetes" WO2008085316
5. "Acyl Bipiperidinyl Compounds as G-protein Coupled Receptor GPR-119 Agonists, Their Preparation, Compositions Containing Such Compounds and Methods of Treatment" WO2008076243
6. "Methods of Performing Cycloadditions, Reaction Mixtures, and Methods of
Performing Asymmetric Catalytic Reactions" US 7230125
C