At the root of cellular biology lie proteins, the conformational dynamics of which dominate dynamic processes such as protein folding, structural plasticity, and functional recognition important for whole-cell function. In these contexts, it has become increasingly clear that static snapshots of proteins are insufficient to unravel their complex role in cellular activity. Instead, the structural and dynamical details of proteins must be measured simultaneously at high spatial and temporal resolution to reveal clear pictures of the dynamics processes. Such a requirement is currently difficult for experimental technologies.
Computer simulations provide the unique capability of revealing both the structural and dynamic details of molecular processes at atomic resolution. As such, the past decade has experienced an accelerated growth in the application of computer simulations to chemical and biological problems. Our research focuses on the development and application of computational methods that provide in-depth atomic insight into the complex conformational dynamics of proteins, with the ultimate goal of making experimentally testable prediction and assisting experimentalists in creating novel approaches to modulate protein conformational dynamics for biomedical applications.
(1) Development of hybrid-resolution models for realistic simulations of biomolecules
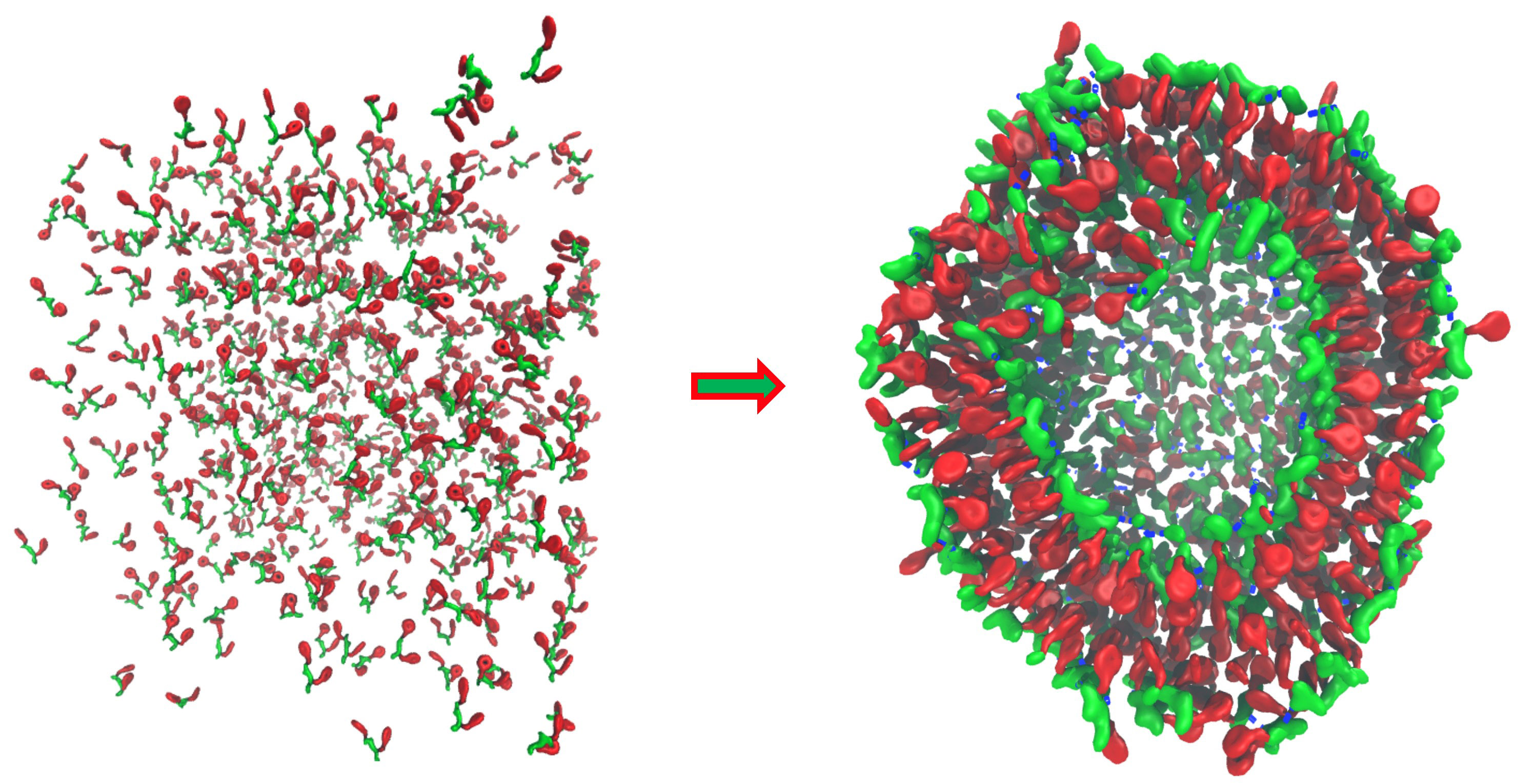
In theory, biomolecular models containing every single atomic detail are the most accurate computational way to probe protein conformational dynamics. In practice, however, due to the expensive computational costs required for simulations with such details, all-atom models are oftentimes applicable only for simulating structural changes on a timescale far shorter than needed for observing biologically relevant events. To simulate processes on much longer timescale, coarse-grained (CG) models are usually employed to reduce spatial resolution and thereby speed up calculations. Due to the loss of atomic detail, however, many CG models are unable to properly model processes involving complicated conformational changes arising in proteins during processes such as folding, aggregation, and protein-protein interactions.
An appealing solution to this problem is to develop a model combining atomistic descriptions of proteins with coarse-grained environments. The model would permit the modeling of protein conformational dynamics with adequate detail but in a highly efficient manner. Our group have conceived and developed one such model, namely PACE (Protein with Atomic detail in Coarse-grained Environment), that enhances simulations significantly as compared to all-atom models. The potential of PACE has been manifested by its application in folding simulations of structurally diversified proteins with an accuracy rivaling that of all-atom models.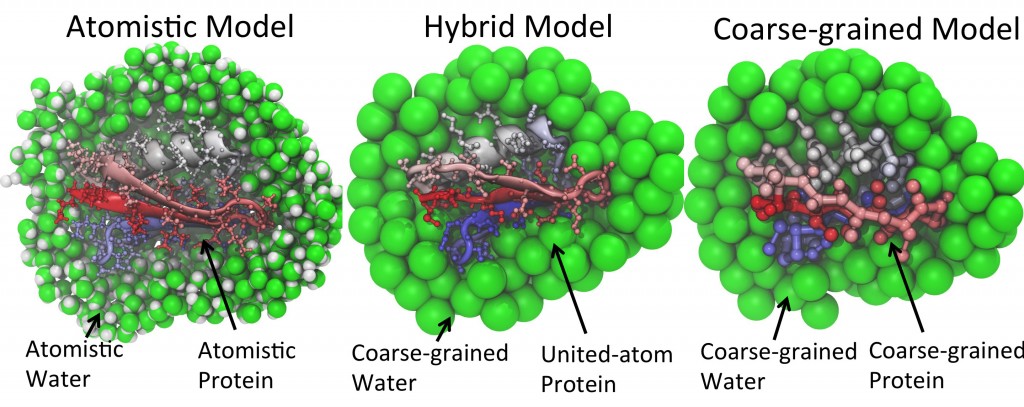
Despite the successful outcomes, the application of PACE is limited to simple protein systems in aqueous environment. In reality, functional proteins operate in a complex environment where they interact constantly with various cellular components. Simulations of such systems need, however, computational methods that can model very large, multiple-component systems on a sufficiently long timescale while still capable of capturing critical details relevant to bio-functions. To meet these requirements, our lab is focusing on the development of next generation hybrid-resolutions model that can be applied to much more realistic simulations of biomolecular systems.
(2) Molecular basis of protein-protein interactions involving intrinsically disordered proteins
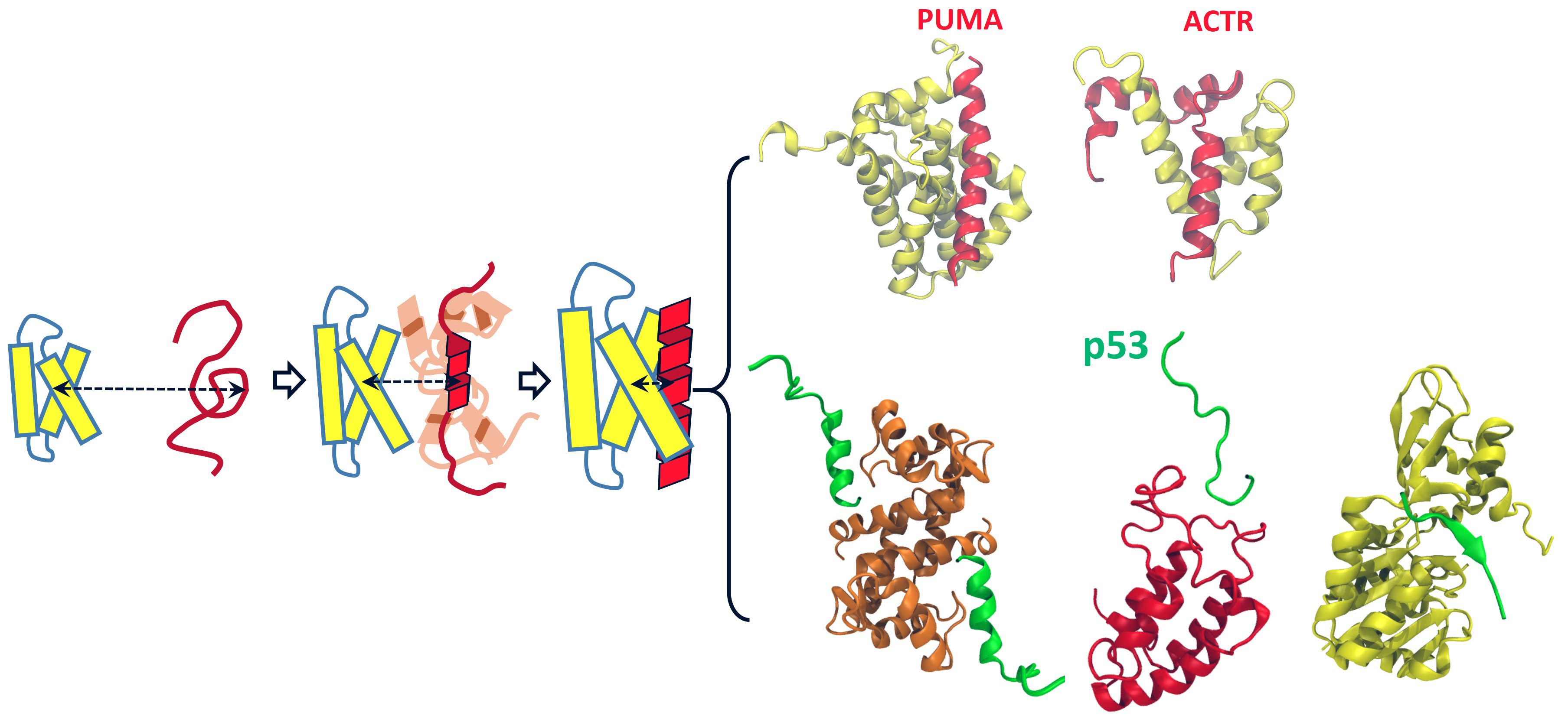
The molecular basis of activities of all living cells, regardless of complexity, arises from underlying networks of interactions between individual proteins. In the networks lie a class of proteins, called intrinsically disordered proteins (IDP), which do not exhibit well-defined structures under native conditions. Despite the lack of structures when isolated, IDPs can recognize and associate with their partners in a fast and specific manner, fulfilling their bio-functions by forming structured complexes. Owning to their structural plasticity, IDPs usually have multiple partners and thus are often found in the hubs of the protein interaction networks, playing a central role in cell signaling and regulation, as manifested by many famous examples such as the role of unstructured parts of p53 proteins in P53’s function as tumor suppressor. Malfunction of many IDPs is linked to serious diseases such as cancers and neurodegenerative diseases. Therefore, it is of both scientific and biomedical significance to understand molecular basis of protein-protein interactions involving IDPs. The key to such understanding is to elucidate how IDPs undergo disorder-to-order structural transition upon complex formation. Our lab is currently developing and applying cutting-edge computational approaches to address critical mechanistic questions regarding the structural transition of IDPs during the course of protein-protein interactions, with a long-term goal of providing new insights for seeking efficient means of modulating or intervening protein-protein interactions for therapeutic purposes.
(3) Mechanism of amyloid formation
Amyloid deposits characterize many diseases, including particularly neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases, in addition to Type II diabetes, each associated with the aggregation of a distinct protein. Protein aggregation is a complex biophysical process, starting with disordered protein monomers in solution, involving a vast number of oligomeric intermediates with diverse structures, and eventually leading to formation of amyloid fibrils in distinct forms. This process arises in environment comprising various cellular components. Such complexity makes extremely difficult to experimentally characterize the atomic detail of each step of the aggregation process. Although computer simulations can characterize structures and dynamics of individual steps of complex processes, their application in protein aggregation is still challenging due to tremendous computational cost needed to simulate the slow process of aggregation. Thus, the details of protein aggregation remain largely unknown. But until these details are filled in, how proteins aggregate will never be fully understood. Nor will the potential be fully developed to exploit protein aggregation as therapeutic targets for amyloid diseases.

Our research focuses on unraveling the mechanism of protein aggregation through computer simulations. To overcome the computational challenge, we are developing a novel computational approach combining multi-scale models, enhanced sampling techniques and statistical mechanics. Our goal is to identify conformational intermediates essential for the aggregation processes. These intermediates, potentially serving as inhibition targets, are currently much needed for inhibitor design aimed at intervening protein aggregation.
(4) Computational design of polypeptide-based materials
Polypeptides are versatile natural building blocks of biological systems, capable of forming, via self-assembly, diverse superstructures with various functions. This unique capability has inspired intense efforts in seeking polypeptide-based molecules that can build from the bottom up materials bearing special structures and, thereby, functions. So far, a number of molecules of this kind have been found to self-assemble into varying superstructures, acting as fabrication templates, absorbing reagents, or catalysts. These identified molecules represent, however, only a tiny fraction of all the possible forms (in terms of peptide sequence and chemical modification) of polypeptide-based molecules. Hence, much work is still needed to explore the better choices of polypeptide-based molecules for material design. Owing to a vast number of possible forms that these molecules can take, a systematic exploration is experimentally prohibited. To overcome this challenge, we are currently developing a highly-efficient, high throughput computational approach that will allow us to computationally examine self-assembly of polypeptide-based molecules in a systematic manner. With this approach, we will identify, through the systematic examination, unknown forms of polypeptide-based molecules that can self-assemble into novel structures and, thus, are worthy of further experimental investigation in seeking unexplored polypeptide-based materials.