News&Event
38.Formal Total Synthesis of N-Methylmaysenine
L. Wang, J. X. Gong, L. J. Deng, Z. Xiang, Z. X. Chen, Y. Wang, J. H. Chen*, Z. Yang*
A novel synthetic approach for the formal total synthesis of N-methylmaysenine (1) has been developed. Key steps involve the Ti-mediated vinylogous Mukaiyama aldol reaction of chiral ketene silyl N,O-acetal with β-dithiane-substituted aldehyde, an aldol condensation, and a ring-closing metathesis reaction.

37.Characterization and Development of Novel Small-Molecules Inhibiting GSK3 and Activating Wnt Signalling
H. B. Zhong, H. X. Zou, M. V. Semenov, D. Moshinsky, X. He, H. Huang, S. Li, J. M. Quan, Z. Yang, S. Lin*
Glycogen synthase kinase 3 (GSK3) is an essential component of the Wnt signaling pathway and plays important roles in regulating cell proliferation, differentiation, and apoptosis. As GSK3 is abnormally upregulated in several diseases including type II diabetes, Alzheimer’s disease and cancer, it has been regarded as a potential drug target. During zebrafish development, inhibition of GSK3 leads to ectopic activation of the Wnt pathway, resulting in a headless embryo. Using this phenotype as an assay we screened a chemical library of 4000 compounds and identified one novel compound, 3F8, which specifically inhibits eye and forebrain formation in zebrafish embryos, resembling a typical Wnt overexpression phenotype. Cell reporterassays, chemical informatics analysis and in vitrokinase experiments revealed that 3F8 is a selective GSK3 inhibitor, which is more potent than SB216763, a commonly used GSK3inhibitor. Based on the structure of 3F8, a new generation of compounds inhibiting GSK3 was synthesized and validated by biological assays. Together, 3F8 and its derivatives could be useful as new reagents and potential therapeutic candidates for GSK3 related diseases.
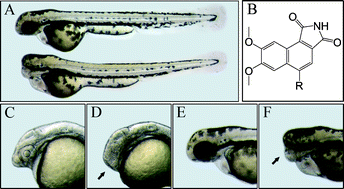
36.Identifying Tumor Cell Growth Inhibitors by Combinatorial Chemistry and Zebrafish Assays
J. Xiang, H. B. Yang, C, Che, H. X. Zou, H. B. Yang, Y. Wei, J. M. H. Zhang*, Z. Yang, S. Lin*
Cyclin-dependent kinases (CDKs) play important roles in regulating cell cycle progression, and altered cell cycles resulting from over-expression or abnormal activation of CDKs observed in many human cancers. As a result, CDKs have become extensive studied targets for developing chemical inhibitors for cancer therapies; however, protein kinases share a highly conserved ATP binding pocket at which most chemical inhibitors bind, therefore, a major challenge in developing kinase inhibitors is achieving target selectivity. To identify cell growth inhibitors with potential applications in cancer therapy, we used an integrated approach that combines one-pot chemical synthesis in a combinatorial manner to generate diversified small molecules with new chemical scaffolds coupled with growth inhibition assay using developing zebrafish embryos. We report the successful identification of a novel lead compound that displays selective inhibitory effects on CDK2 activity, cancer cell proliferation, and tumor progression in vivo. Our approaches should have general applications in developing cell proliferation inhibitors using an efficient combinatorial chemical genetic method and integrated biological assays. The novel cell growth inhibitor we identified should have potential as a cancer therapeutic agent.
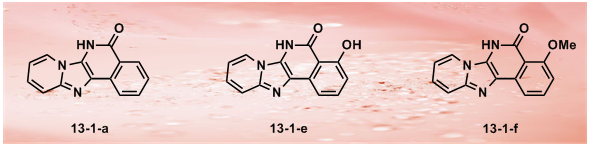
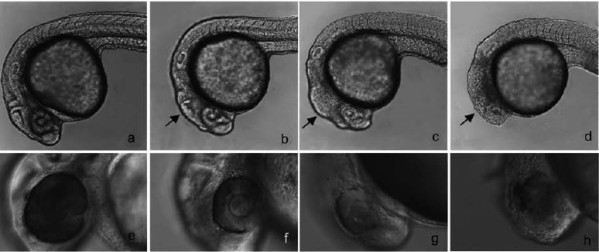
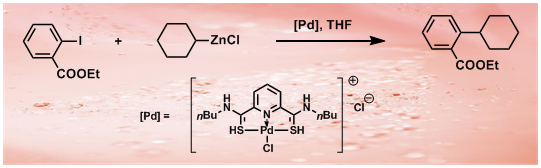
35. Pincer Thioamide and Pincer Thioimide Palladium Complexes Catalyze Highly Efficient Negishi Coupling of Primary and Secondary Alkyl Zinc Reagents at Room Temperature
H. B. Wang, J. Liu, Y. Deng, T. Y. Min, G. X. Yu, X. J. Xu, Z. Yang, A. W. Lei*
Chem. Eur. J. 2009, 15, 1499-1507.
Pincer thioamide PdII complex 2 was prepared, and its reaction with cyclohexylzinc chloride yielded novel pincer thioimide PdII complex 3 besides Pd0 species. The structures of complexes 2 and 3 were confirmed by X-ray analysis. Both complexes are efficient catalysts for Negishi couplings involving primary and secondary alkyl zinc reagents bearing β-hydrogen atoms. At a concentration of 0.1–0.5 mol % both catalysts readily promoted reactions at room temperature or even at 0 °C. The operational simplicity of these processes, in conjunction with the easy accessibility of both catalysts and substrates, promises synthetic utility of this new methodology. An experiment on a scale of 19.35 g carried out at very low catalyst loading of 2 (turnover number: 6 100 000) highlighted the potential application of the catalytic system. Monoalkyl and dialkyl zinc reagents displayed different reactivities and selectivities in reactions with aryl iodides catalyzed by complexes 2 or 3, and isomerization in reactions involving acyclic secondary alkyl zinc derivatives was suppressed by using appropriate amounts of dialkyl zinc reagents. Based on preliminary kinetic profiles and reaction evidence, three possible pathways are proposed for the reactions involving acyclic secondary alkyl zinc reagents to rationalize the difference between mono-alkyl and dialkyl zinc derivatives.
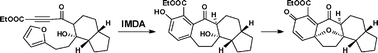
34.A model study for the concise construction of the oxapentacyclic core of cortistatins through intramolecular Diels-Alder and oxidative dearomatization-cyclization reactions
L. Z. Liu, Y. X. Gao, C. Che, N. Wu, D. Z. Wang, C. C. Li*, Z. Yang*
A unified strategy towards the facile construction of the [6.7.6.5] oxapentacyclic skeleton of cortistatins is reported, featuring intramolecular Diels–Alder (IMDA) and oxidative dearomatization–cyclization reactions as key steps.