Publications
152.Navigating the Pauson–Khand Reaction in Total Syntheses of Complex Natural Products

Zhen Yang*
Acc. Chem. Res. 2021, 54(3), 556–568
“Total synthesis endeavors provide wonderful opportunities to discover and invent new synthetic reactions as a means to advance organic synthesis in general. Such discoveries and inventions can occur when the practitioner faces intransigent problems that cannot be solved by known methods and/or when method improvements are desired in terms of elegance, efficiency, cost-effectiveness, practicality, or environmental friendliness” (K. C. Nicolaou et al. from their review in CCS Chem.2019, 1, 3–37). To date tens of thousands of bioactive compounds have been isolated from plants, microbes, marine invertebrates, and other sources. These chemical structures have been studied by chemists who scanned the breadth of natural diversity toward drug discovery efforts. Drug-likeness of natural products often possesses common features including molecular complexity, protein-binding ability, structural rigidity, and three-dimensionality. Considering certain biologically important natural products are scarce from natural supply, total synthesis may provide an alternative solution to generating these compounds and their derivatives for the purpose of probing their biological functions. Natural products bearing quaternary carbon stereocenters represent a group of biologically important natural entities that are lead compounds in the development of pharmacological agents and biological probes. However, the stereocontrolled introduction of quaternary carbons, with vicinal patterns that substantially expand the complexity of molecular architectures and chemical space in particular, presents distinct challenges because of the high steric repulsion between substituents. Though remarkable advance has been seen for quaternary carbon stereocenter generation, the process remains a daunting challenge given that the formation of highly congested stereocenters increases the difficulty in achieving orbital overlap.
In the past two decades, our group has initiated a program to develop synthetic strategies and methods with the aim of advancing the frontiers of the total syntheses of biologically important complex natural products bearing all-carbon quaternary stereogenic centers. Typical endeavors have involved the use of a Pauson–Khand (PK) reaction as a key step in constructing core structures with all-carbon quaternary stereogenic center(s), with the aid of well-orchestrated thiourea–Co- and thiourea–Pd-catalyzed PK reactions. These methodological advances have enabled us to achieve total syntheses of a series of topologically complex natural products with diverse structural features. These methods will enable the assembly of molecules with improved biological functions and provide tool compounds for elucidation of mechanism of action or identification of potential cellular targets.

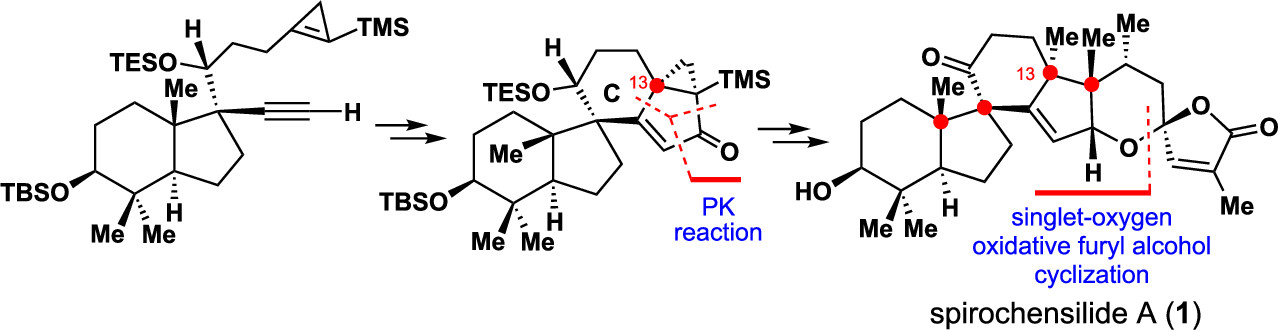
151.Asymmetric Total Synthesis of (−)-Spirochensilide A, Part 2: The Final Phase and Completion

Xin-Ting Liang, Bao-Chuan Sun, Nan Zhang, Zhong-Chao Zhang, Yuan-He Li, Qian-Qian Xu, Chang Liu, Jia-Hua Chen,* and Zhen Yang*
J. Org. Chem. 2021, 86(3), 2158–2172
The final phase of the total synthesis of (−)-spirochensilide A is described. A tungsten-mediated cyclopropene-based Pauson–Khand reaction was developed to form the spiral CD ring system with desired stereochemistry at the C13 quaternary center. Other important steps enabling completion of this synthesis included an intermolecular aldol condensation to link the ABCD core with the EF fragment and a Cu-mediated 1,4-addition to stereoselectively install the C21 stereogenic center. The chemistry developed for this total synthesis of (−)-spirochensilide A (1) will aid the synthesis of polycyclic natural products bearing this unique spiral ring system.
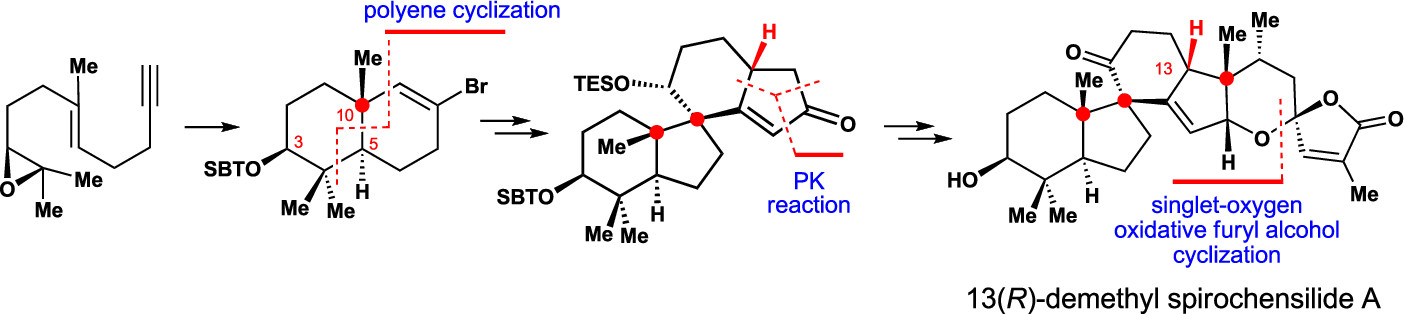
150.Asymmetric Total Synthesis of (−)-Spirochensilide A, Part 1: Diastereoselective Synthesis of the ABCD Ring and Stereoselective Total Synthesis of 13(R)-Demethyl Spirochensilide A
Xin-Ting Liang, Bao-Chuan Sun, Chang Liu, Yuan-He Li, Nan Zhang, Qian-Qian Xu, Zhong-Chao Zhang, Yi-Xin Han, Jia-Hua Chen,* and Zhen Yang*
J. Org. Chem. 2021, 86(3), 2135–2157
A concise and diastereoselective construction of the ABCD ring system of spirochensilide A is described. The key steps of this synthesis are a semipinacol rearrangement reaction to stereoselectively construct the AB ring system bearing two vicinal quaternary chiral centers and a Co-mediated Pauson–Khand reaction to form the spiro-based bicyclic CD ring system. This chemistry leads to the stereoselective synthesis of 13(R)-demethyl spirochensilide A, paving the way for the first asymmetric total synthesis of (−)-spirochensilide A.
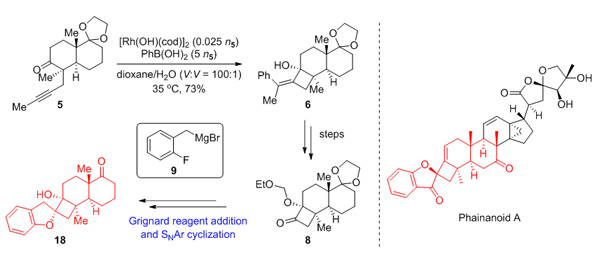
149.Synthetic Study Toward the 4,5-Spirocycle Skeleton of Phainanoids

Jiang, Chongguo†, Chen, Sijia†, Gong, Jianxian*, Yang, Zhen*
Acta Chim. Sinica 2020, 78(9), 928—932
Attempts to synthesize the 4,5-spirocycle skeleton of Phainanoids by rhodium-catalyzed arylative cyclization of alkynone 5 and the addition of Grignard reagent 9 to α-alkoxyl cyclobutone 8, followed by intramolecular SNAr reaction are reported. Phainanoids, highly modified triterpenoids, were isolated from Phyllanthus hainanensis by Yue and co-workers. They have been found to show intriguing immunosuppressive activities. The most potent of them, Phainanoid F, inhibit the proliferation of T cells with an IC50 value of (2.04±0.01) nmol/L and B cells with an IC50 value <(1.60±0.01) nmol/L. The noteworthy activities and the lack of Phainanoids in nature resources make the synthesis of them for further biological evaluation a challenge for chemists. Our synthesis started from known compound 1, after Birch reduction and alkylation to give alkynone 5. The rhodium-catalyzed arylative cyclization of alkynone 5 to deliver tetrasubstituted cyclobutene 6 was performed by the following procedure. Under an atmosphere of Ar, to an oven-dried Schlenk tube with[Rh(OH)(cod)]2 (35.5 mg, 0.078 mmol, 0.012n5), phenylboronic acid (2.0 g, 16.3 mmol, 2.5n5), were added a solution of ketone 5 (1.9 g, 6.5 mmol, 1.0n5) in 1,4-dioxane (32.0 mL) and H2O (0.3 mL) at room temperature. The mixture was stirred at 35℃ for 12 h. Another [Rh(OH)(cod)]2 (35.5 mg, 0.078 mmol, 0.012n5) and phenylboronic acid (2.0 g, 16.3 mmol, 2.5n5) was added to the mixture. The mixture was stirred at 35℃ for 12 h. Subsequently, hydroxyl group was protected with ethoxymethyl (EOM) group to furnish 7, followed by ozonolysis to generate ketone 8. Ketone 8 was reacted with fresh prepared Grignard reagent 9 in Felkin-Anh modelinstead ofthe Cram’s chelation-control model to deliver alcohol 10. The explanation of the diastereoselectivity of this reaction could be illustrated from two aspects:(1) the rigid structure of α-alkoxyl cyclobutone 8 increased the energy barrier for the transition state of chelation between magnesium ions and alkoxyl substituent; (2) the magnesium ions were not chelated with the alkoxyl substituent as well as the carbonyl oxygen was due to the intramolecular chelation with fluorine atom. Alcohol 10 underwent intramolecular SNAr reaction and deprotection to deliver 4,5-spirocycle compound 18.
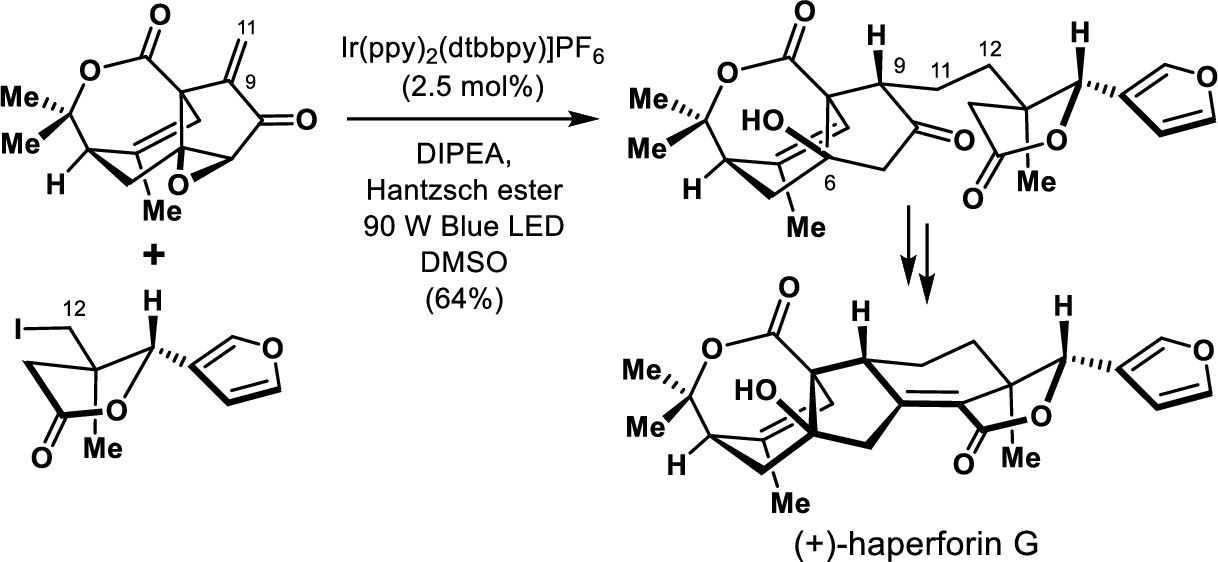
148.Total Synthesis of (+)-Haperforin G

Wei Zhang, Zhenyu Zhang, Jun-Chen Tang, Jin-Teng Che, Hao-Yu Zhang, Jia-Hua Chen,* and Zhen Yang*
J. Am. Chem. Soc. 2020, 142(46), 19487–19492
A concise chemical synthesis of (+)-haperforin G in 20 steps from commercially available starting materials is achieved with the integration of the Co-catalyzed intramolecular Pauson–Khand reaction for the stereoselective construction of cyclopentanone bearing an all-carbon quaternary stereogenic center at the bridge-head position and the light-initiated photocatalysis for convergent and asymmetric cross-coupling of the unstabilized C(sp3)-radical with an enone. The developed chemistry paves the way to synthesizing structurally diverse analogs of haperforin G (6).